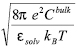![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Electrostatic potential calculation for biomolecules - creating a database of pre-calculated values reported on a per residue basis for all PDB protein structures W. Rocchia1 and G. Neshich2
Genet. Mol. Res. 6 (4): 923-936 (2007) ABSTRACT. STING and JavaProtein Dossier provide a collection of physical-chemical parameters, describing protein structure, stability, function, and interaction, considered one of the most comprehensive among the available protein databases of similar type. Particular attention in STING is paid to the electrostatic potential. It makes use of DelPhi, a well-known tool that calculates this physical-chemical quantity for biomolecules by solving the Poisson Boltzmann equation. In this paper, we describe a modification to the DelPhi program aimed at integrating it within the STING environment. We also outline how the "amino acid electrostatic potential" and the "surface amino acid electrostatic potential" are calculated (over all Protein Data Bank (PDB) content) and how the corresponding values are made searchable in STING_DB. In addition, we show that the STING and JavaProtein Dossier are also capable of providing these particular parameter values for the analysis of protein structures modeled in computers or being experimentally solved, but not yet deposited in the PDB. Furthermore, we compare the calculated electrostatic potential values obtained by using the earlier version of DelPhi and those by STING, for the biologically relevant case of lysozyme-antibody interaction. Finally, we describe the STING capacity to make queries (at both residue and atomic levels) across the whole PDB, by looking at a specific case where the electrostatic potential parameter plays a crucial role in terms of a particular protein function, such as ligand binding. BlueStar STING is available at http://www.cbi.cnptia.embrapa.br. Key words: Electrostatic potential of biomolecules, DelPhi, STING, Protein structure analysis INTRODUCTION JavaProtein Dossier (JPD; Neshich et al., 2004) is a new concept database and visualization tool for both structures deposited at the Protein Data Bank (PDB; Berman et al., 2000) and those derived either by models or experiments, but not yet available from PDB. JPD is a part of the STING (Neshich et al., 2003, 2005a) environment which provides one of the most comprehensive collections (Neshich et al., 2005b) of physical-chemical parameters describing protein sequence, structure, stability, function, and interaction with other macromolecules. Electrostatics is believed to play a pivotal role in regulating interactions between biological macromolecules, such as proteins and nucleic acids (Radic et al., 1997; Sheinerman et al., 2000). Among other aspects, the electrostatic contribution to the (de)solvation process has proved to be markedly important in many biological phenomena. For many applications, that contribution is modeled as a dielectric linear response to the electric field generated by the charge borne by the biomolecular system. Consistent with this model, the Poisson-Boltzmann equation (PBE) has proved to be able to provide quantitative estimates of the electrostatic interaction energy of biomolecules (Honig and Nicholls, 1995). In addition to 311 other descriptors, JPD encompasses also the description of some electrostatic features, providing the numerical value of the mean electrostatic potential (EP) at each residue and at some relevant atoms, as well as the potential over the molecular surface, as described in what follows. Poisson-Boltzmann equation and biomolecules - STING variation The EP is calculated by solving the PBE within a finite difference scheme by using the DelPhi program (Columbia University) with a few ad hoc modifications. PBE is used in the context of the continuum approximation of matter (Cramer and Truhlar, 1999). In this model, molecules and solvent are treated as media that react linearly and uniformly to the electrostatic field generated by some kind of source, typically the charge distribution on the molecules themselves. The detail of the description of the solute molecules is greater than the one of the solvent. Biomolecules are described at the atomic level, where each atom has its own partial charge, located in its center, and radius. Usually, but since the latest version of DelPhi this is no longer mandatory (Rocchia et al., 2001), all atoms belonging to a biomolecule have the same, low, dielectric constant, that is, they react with the same intensity to a preexisting electric field. The Connolly-Richards molecular surface (Connolly, 1983; Eisenhaber et al., 1995; Rocchia et al., 2002) is then built, being the surface generated by a solvent molecule that rolls over the solute. The space volume outside the molecular surface is treated as a uniform, high dielectric medium, with a typical dielectric constant value of 80. A suitable space-filling routine (Eisenhaber et al., 1995) assigns the remaining space regions to the dielectric constant of the closest solute atom. Usually, the concept of dielectric constant is used to describe the collective behavior of matter and does not go down to the atomic level of description. However, in this context, dielectric constant is used to describe any linear response of the medium that has not been modeled explicitly. For example, covalent bonds cause a charge imbalance which is already taken into account explicitly in the partial charges, and the dielectric constant should not account for this. In contrast, some researchers are interested in getting a quick idea of what is the effect of exposed mobile side chains, and they prefer to use a large dielectric constant for these side chains rather than averaging over the different conformers of the molecule. By default, the dielectric constant of solute molecules is set to be equal to 2, accounting for electronic polarizability. Once the system is physically modeled, the PBE is solved according to a finite difference scheme. Finite difference solutions to the PBE involve solving the equation in a lattice representation (Holst et al., 2000). This representation implies that charges and dielectric properties of the system are mapped on the lattice, that is, an ordered, in our case cubic, ensemble of so-called grid points where the EP is calculated. In the model, charge is mapped on the grid points closest to the atom center according to a tri-linear interpolation scheme (Rocchia, 2005). The dielectric constant, which is more generally a property of the "medium", in turn is mapped on the mid-points (a mid-point is the central point between two adjacent grid points). In order to make it possible to handle different dielectric regions, each mid-point has to be associated with the dielectric constant of the region in which it is located. An important task in the mapping phase is the correct assignment of boundary grid points, located on the interface between different media. In case of a low-charged system, PBE is usually linearized, leading to a simpler numerical problem. For a solution of a symmetric monovalent salt, the linearized PBE has the following form:
where
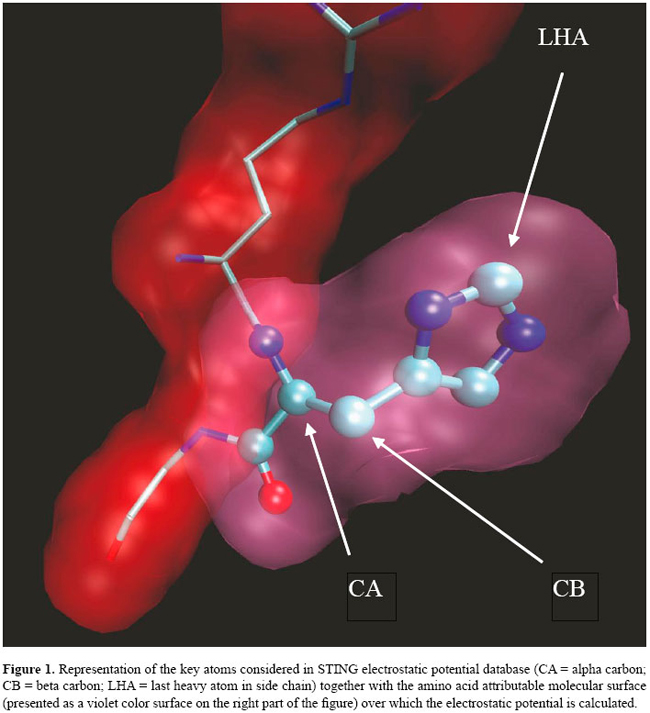
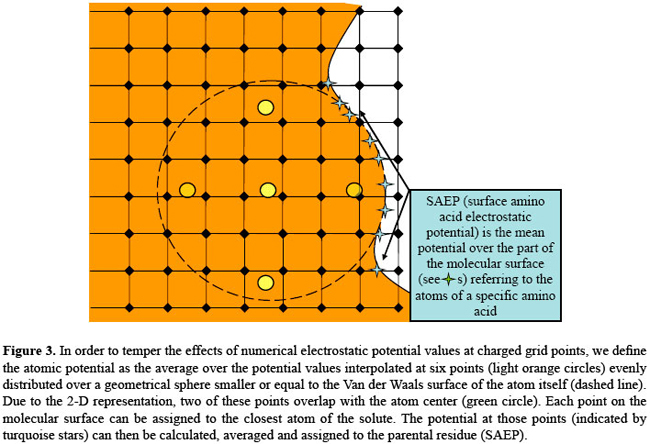
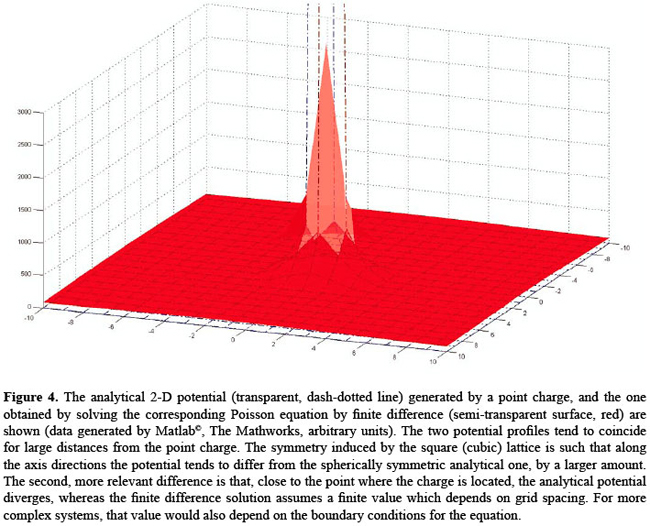
if The above form of the equation is sometimes referred to as the strong form, in that the solving potential is required to be twice differentiable for the equation to hold in the traditional sense. The finite differences discretization process passes through the so-called weak formulation (Richards, 1977), which is less strict as far as the conditions on the derivatives of the solution are concerned. Mapping the system on a lattice has some consequences, common to all of the finite difference schemes, of which the thorough user should be aware. First of all, the solution of the weak form nears the solution of the original equation only far away from the charged grid points. The grid potential at a charged site not only differs from the "strong" solution, where it should diverge, but its value depends quite strongly on the grid spacing and on the system position relative to the lattice. Therefore, shifting the system of fractions of Angstroms, as well as changing resolution, may very well result in quite different potential values at charged grid points. In the light of this reasoning, if the user wants to obtain potential values referring to individual atoms, it is advisable to use in the simulation a grid spacing that is a fraction of the smallest atom-to-surface distance in the molecular system (usually a value of 0.4 Å can be acceptable). Furthermore, as we already stated, the EP at each atom center is affected by the inaccuracy due to space discretization. One possible way to derive a more reliable atomic potential (AP) would be to average over many different system-to-lattice relative positions, but this would be very time-consuming; the strategy we adopt here is to define the AP as the average potential calculated at six points evenly distributed on the Van der Waals surface of the atom itself, or at a user-defined distance. In this way the contribution of the grid potential values at charged sites is reduced and the results are more reproducible. Given our definition of AP, two quantities of interest can be further introduced: the amino acid EP and the surface amino acid EP. The amino acid EP is calculated as an average of the AP over each amino acid (Figure 1). From a numerical viewpoint, this average is closer to the value obtained by a numerical solver at a lower resolution, such as GRASP (Nicholls et al., 1991). The rationale for this is that when the grid spacing is large (Figure 2), several partial charges happen to be mapped on the same grid point, the net result looking very much like a spatial averaging. The suface amino acid EP (Figure 3) is the average potential over the part of the molecular surface contributed by a given amino acid. Its derivation is somehow more complicated. The PBE solver that we used, i.e., DelPhi, has a routine that iteratively calculates the above-mentioned Connolly-Richards molecular surface (Connolly, 1983; Eisenhaber et al., 1995; Rocchia et al., 2002); the importance of this surface resides in the fact that the solvation contribution, better known as the reaction field energy, is generated by the polarization charge arising at the boundary between the molecule and the solvent. Therefore, all over the molecular surface there are points where the polarization charge is calculated, each of these points can be referred to an atom and, therefore, to a residue. Furthermore, it is important to note that, in this model, EP is a continuous variable across the molecular surface. Our claim of higher reliability is based on this last remark and on the fact that it is very unlikely to use PBE numerical solution at charged lattice points to calculate EP at the molecular surface.
The innovative character of this approach resides in the fact that prior to the modifications we introduced into DelPhi, and then integrated into the STING environment, it was only possible to have EP reported at atom centers, with the inherent inaccuracy arising from the fact that most of the times a charge localizes in those positions (see Figure 4). In contrast, our novel definition of atomic potential virtually involves no charged grid points, leading to a higher reproducibility of the results. Furthermore, before the new features were introduced, there was no automatic way to link a point lying on the molecular surface to its parent atom. Thus, the task of calculating the potential of the solvent-exposed part of a specific residue has been now made much easier. In addition, JPD also reports the EP values calculated and attributed to the alpha carbon atom and the last heavy atom (LHA) in the side chain of each amino acid.
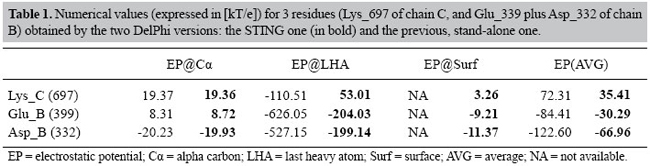
High throughput procedure for calculating electrostatic potential in STING The STING script for calculation of EP values over the entire PDB starts with the IRIX OS version of the Reduce program (Word et al., 1999), which adds hydrogen atoms to the protein. Default values for the flags given to the program are: -HIS -NOADJust. Subsequently, the multi-chain proteins are processed so that the chains are separated and stored in temporary files. The DelPhi program is then run for each separate chain. The output files are stored, indexed by pdb id and chain id. Default parameters for the DelPhi run are: percentage filling = 80%, grid spacing = 0.4 Å, null ionic strength, probe radius = 1.4 Å, Stern layer width = 2.0 Å, internal relative dielectric constant = 2, and solvent relative dielectric constant = 80. For definition of the atomic electrostatic potential, a distance of 0.6 Å from the atomic center was chosen. Comparative studies of calculated values for electrostatic potential in STING and DelPhi In order to better represent the novelty of data stored in STING_DB relative to EP, we decided to compare the interesting quantities from the same source (namely: 1nmd PDB code, chains: B and C, residues: C_697, B_399 and B_332, atoms: Cα, Cβ and LHA) but derived using the two different versions of the DelPhi program: before and after modification (Table 1).
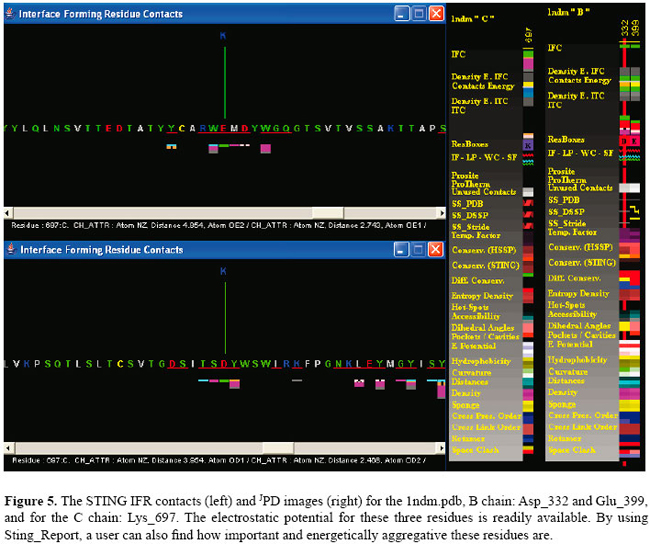
As the data clearly show, the EP values show the same tendency, except for two important aspects: a) the LHA EP for the Lys_697 shows a very different behavior in the two calculations. One can clearly see that the earlier version of DelPhi provides EP values which are much more in accordance with the charge values than the STING version, where the environment EP plays a major corrective role. b) Surface values referring to single amino acids are not easily obtainable from the earlier DelPhi version. An obvious advantage of having pre-calculated values is that the user does not need to overcome the learning barrier of using DelPhi, and he/she can just read data already available in STING_DB, reported in residue by residue fashion. Electrostatic potential at the antigen-antibody interface analyzed with the STING electrostatic potential It was reported in the literature (Sinha et al., 2002) that some of the networked salt bridges present in HH26-HEL stand as very strong and play a major role in stabilizing the molecular system. We focused on interface contacts and electrostatic contributions toward binding. In general, EP factor, beyond helping binding, also limits the flexibility of the system, making the interface geometry very rigid. Therefore, we decided to use this particular case as a biologically interesting example where to apply our analysis. The PDB file 1nmd.pdb was used to test how STING can perform in terms of how quickly a user can extract the information relative to the EP which is stored in the STING_DB. In Figure 5, we show the information produced by the STING IFR contact module, showing the attractive electrostatic interaction between the Lys_697 of the C chain (lysozyme) and both the Asp_332 (the bottom left inset) and Glu_399 (the upper left inset) of the B chain (antibody). On the right side of the same figure, we show the JPD content for the Lys_697 of the C chain and next to it, on the right side, the content concerning two negatively charged residues of the B chain. The variation of the colors indicates existence of 4 values in the electrostatic potential parameter line. If a user holds a mouse over this line, the numerical values for EP STING parameter show up (see Table 1, bold values).
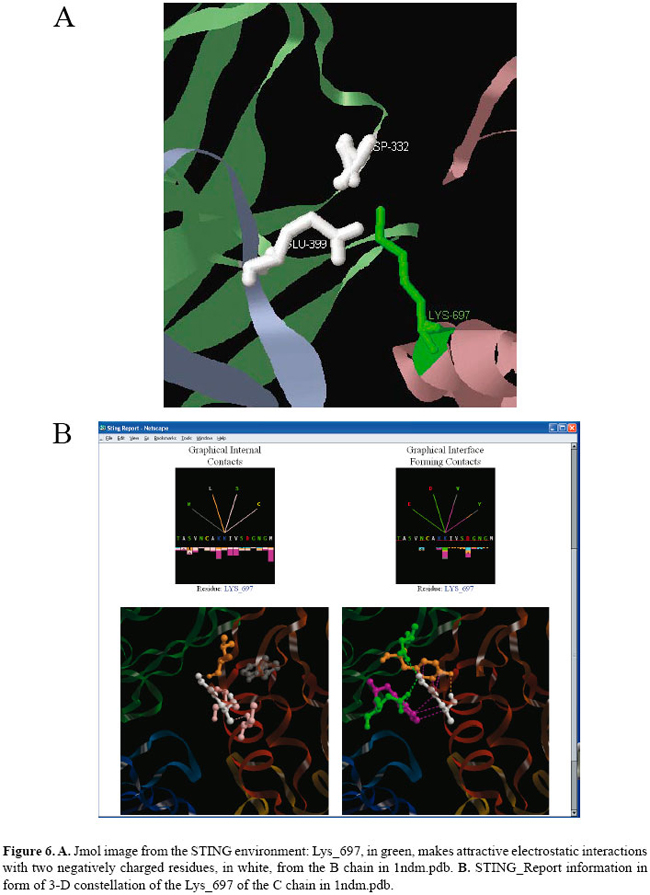
According to the literature, those residues were shown to produce the largest EP contribution among all residues at the interface. Indeed, if we look at the STING_Report for the residue Lys_C (697) (not shown here) we can find that this residue is responsible for all salt bridges across the interface, and this is the largest interaction energy contribution. Figure 6A presents the 3-D constellation of Lys_697 (in green) against the two negatively charged residues across the interface (in white color). In Figure 6B, we show both internal and interface contacts for Lys_697. Example of a STING_DB query involving electrostatic potential values A relational database query is a question about data, and the answer consists of a new relation containing the result. For example, we may want to find the ensemble of structures, selected from the whole PDB, that has the following characteristics: a) all structures that have a pocket (i.e., a depression in a protein surface, generally accessible to the solvent over its entire extent) with a volume ranging between 200 and 300 Å, b) the residues forming such pocket would be highly conserved (with relative entropy, as calculated by the STING SH2Qs (Higa et al., 2006), being lower than 20), and c) the residues forming such pocket would also have an EP value for the surface portion attributable to that particular amino acid, positive or higher than 30 kT/e.
The biological meaning of this query (also described in Figure 7) would be to retrieve an ensemble of selected structures that could bind a negatively charged ligand. Clearly, such a search is possible due to the availability of pre-calculated data relative to EP. They were first stored in STING_DB, and then, the flat (text) file was imported in a relational data base (STING_RDB) from which complex queries can be made.
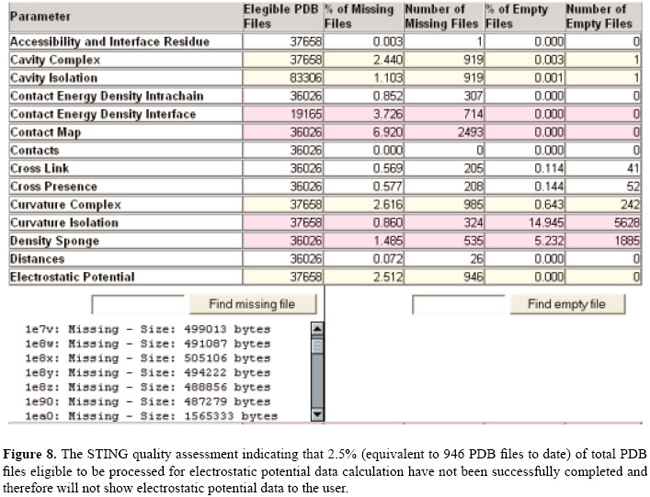
Check for electrostatic potential data in STING_DB for a given PDB file In order to provide the user with the capability of verifying whether a given PDB structure was successfully processed during the high throughput STING_DB update, we introduced in the STING version "Star" (Neshich et al., 2006), the quality assessment (QA) module. Consequently, if the user encounters a highly unlikely case where the EP data are non-existent for a given PDB structure, he/she may first opt to consult STING QA and to verify if that particular structure is included in the list of PDB files for which the procedure we implemented to calculate the four EP parameters could not be successfully completed. The reasons for such outcome (incomplete run for calculation of EP) may be various, most likely due to inconsistencies in nomenclature of atoms used by the authors of the deposited structure. In Figure 8, we show the current (July, 2006) status of the QA module, reporting the percentage of the total number of PDB files that were found either empty (file created but no data calculated) or where the procedure was aborted during EP calculation (for unknown reasons) and no file was created.
CONCLUSIONS The EP is considered as one of the key factors influencing protein interactions. In the STING relational database, which is composed of more than 300 structural parameters concerning protein analysis, special attention was paid to the calculation of EP. Its value is calculated on a per atom basis and then reported for all eligible PDB files in a residue by residue fashion. Four pre-calculated categories are shown: 1) EP at the alpha carbon atom, 2) EP value at the LHA atom, 3) average EP value over all amino acid atoms, and 4) EP value averaged over the patch of the molecular surface that is attributable to that particular amino acid. The main features that we outlined in this study can be summarized as follows:
Apart from the above-mentioned features, STING_RDB, together with its EP element, allows biologically relevant questions to be easily addressed now. We presented one test case in which an antibody-lysozyme interaction was analyzed from the platform of pre-calculated data available within STING_DB. FUTURE DEVELOPMENTS We expect to further improve both the quality of STING_DB and its EP element, by constantly decreasing the percentage of PDB files for which our procedure has failed to generate usable output, as well as trying to improve the speed of the procedure and the algorithmic aspect in terms of obtaining more reliable data. Concerning EP calculation, particular attention needs to be paid to PDB files presenting non-standard residues or nomenclature, which could hinder correct radius and charge assignment to the structure. ACKNOWLEDGMENTS We would like to thank the Department of Pharmaceutical Sciences, University of Bologna, Italy, for the use of Matlab software for data analysis. FIRB project ‘‘Laboratorio Nazionale sulle Nanotecnologie per Genomica e Postgenomica (NG-Lab)’’ is gratefully acknowledged for the financial support. REFERENCES Berman HM, Westbrook J, Feng Z, Gilliland G, et al. (2000). The protein data bank. Nucleic Acids Res. 28: 235-242. Connolly ML (1983). Solvent-accessible surfaces of proteins and nucleic acids. Science 221: 709-713. Cramer CJ and Truhlar DG (1999). Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem. Ver. 99: 2161-2200. Eisenhaber F, Lijnzaad P, Argos P, Sander C, et al. (1995). The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 16: 273-285. Higa RH, Cruz SA, Kuser PR, Yamagishi ME, et al. (2006). Building multiple sequence alignments with a flavor of HSSP alignments. Genet. Mol. Res. 5: 127-137. Holst M, Baker N and Wang M (2000). Adaptive multilevel finite element solution of the Poisson-Boltzmann equation I: Algorithm and examples. J. Comput. Chem. 21: 1319-1342. Honig B and Nicholls A (1995). Classical electrostatics in biology and chemistry. Science 268: 1144-1149. Neshich G, Togawa RC, Mancini AL, Kuser PR, et al. (2003). STING Millennium: a web-based suite of programs for comprehensive and simultaneous analysis of protein structure and sequence. Nucleic Acids Res. 31: 3386-3392. Neshich G, Rocchia W, Mancini AL, Yamagishi ME, et al. (2004). JavaProtein Dossier: a novel web-based data visualization tool for comprehensive analysis of protein structure. Nucleic Acids Res. 32: W595-W601. Neshich G, Borro LC, Higa RH, Kuser PR, et al. (2005a). The Diamond STING server. Nucleic Acids Res. 33: W29-W35. Neshich G, Mancini AL, Yamagishi ME, Kuser PR, et al. (2005b). STING Report: convenient web-based application for graphic and tabular presentations of protein sequence, structure and function descriptors from the STING database. Nucleic Acids Res. 33: D269-D274. Neshich G, Mazoni I, Oliveira SR, Yamagishi ME, et al. (2006). The Star STING server: a multiplatform environment for protein structure analysis. Genet. Mol. Res. 5: 717-722. Nicholls A, Sharp KA and Honig B (1991). Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281-296. Radic Z, Kirchhoff PD, Quinn DM, McCammon JA, et al. (1997). Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin. J. Biol. Chem. 272: 23265-23277. Richards FM (1977). Areas, volumes, packing and protein structure. Annu. Rev. Biophys. Bioeng. 6: 151-176. Rocchia W (2005). Poisson-Boltzmann equation boundary conditions for biological applications. Math. Comput. Model. 41: 1109-1118. Rocchia W, Alexov E and Honig B (2001). Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B. 105: 6507-6514. Rocchia W, Sridharan S, Nicholls A, Alexov E, et al. (2002). Rapid grid-based construction of the molecular surface for both molecules and geometric objects: applications to the finite difference Poisson-Boltzmann method. J. Comput. Chem. 23: 128-137. Sheinerman FB, Norel R and Honig B (2000). Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol. 10: 153-159. Sinha N, Mohan S, Lipschultz CA and Smith-Gill SJ (2002). Differences in electrostatic properties at antibody-antigen binding sites: implications for specificity and cross-reactivity. Biophys. J. 83: 2946-2968. Word JM, Lovell SC, Richardson JS and Richardson DC (1999). Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285: 1735-1747. |
|
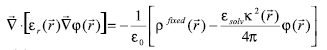
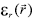 is the local relative dielectric constant
is the local relative dielectric constant  sulv, is that of the solvent,
sulv, is that of the solvent,