![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
Sting_RDB: a relational database of structural parameters for protein analysis with support for data warehousing and data mining S.R.M. Oliveira, G.V. Almeida, K.R.R. Souza, D.N. Rodrigues, P.R. Kuser-Falcão, Genet. Mol. Res. 6 (4): 911-922 (2007) ABSTRACT. An effective strategy for managing protein databases is to provide mechanisms to transform raw data into consistent, accurate and reliable information. Such mechanisms will greatly reduce operational inefficiencies and improve one’s ability to better handle scientific objectives and interpret the research results. To achieve this challenging goal for the STING project, we introduce Sting_RDB, a relational database of structural parameters for protein analysis with support for data warehousing and data mining. In this article, we highlight the main features of Sting_RDB and show how a user can explore it for efficient and biologically relevant queries. Considering its importance for molecular biologists, effort has been made to advance Sting_RDB toward data quality assessment. To the best of our knowledge, Sting_RDB is one of the most comprehensive data repositories for protein analysis, now also capable of providing its users with a data quality indicator. This paper differs from our previous study in many aspects. First, we introduce Sting_RDB, a relational database with mechanisms for efficient and relevant queries using SQL. Sting_rdb evolved from the earlier, text (flat file)-based database, in which data consistency and integrity was not guaranteed. Second, we provide support for data warehousing and mining. Third, the data quality indicator was introduced. Finally and probably most importantly, complex queries that could not be posed on a text-based database, are now easily implemented. Further details are accessible at the Sting_RDB demo web page: http://www.cbi.cnptia.embrapa.br/StingRDB. Key words: Sting database, Protein structure analysis, Data warehousing, Data mining, Data mart INTRODUCTION A protein database is a tool that can help biologists store, manage, disseminate, and understand information related to protein sequence/structure/function/stability and its complex network of interactions with other molecules in biochemical pathways. One of the common characteristics of protein databases is that they are often noisy, "high-dimensional", sparse, and redundant (Radivojac et al., 2004). In general, three sources contribute to the noise in protein data: i) biological complexity and variability (e.g., protein modification upon transcription related to organism, gender or tissue-specific cell characteristics, etc.); ii) limitations of experimental procedures such as sample preparation protocols and techniques (e.g., X-ray crystallography or NMR spectroscopy), and iii) human error related to laboratory conditions, misinterpretation of results, database labeling and curation. High dimensionality and sparseness of protein databases are often consequences of so-called orthogonal (binary) data representation (Qian and Sejnowski, 1988) which is predominantly used in this area. Each locus in a protein is represented by a 20-bit vector in which the observed amino acid is represented by a one and the remaining amino acids are represented by zeros (e.g., for alanine the representation is 100000000000000000000). As a result, orthogonal data representation produces a high-dimensional sample with 20 features, 19 of which are zeros. It also introduces noise since in such a representation long-range sequence interactions are ignored. Another important characteristic of protein datasets is its high redundancy (Berman et al., 2000; Dunker et al., 2002). For example, many proteins correspond to different states or are engineered to facilitate lab experiments (mutants). Such proteins may easily form a large body of redundant data, which can lead to unrealistically high estimates of results (bias). Clearly, an effective strategy for managing protein databases is to provide a means to transform raw data into consistent, accurate and reliable information. In doing so, operational inefficiencies are greatly reduced, and our ability to better handle scientific objectives and interpret the research results will be improved significantly. To achieve this challenging goal, we introduce here Sting_RDB, a relational database of structural parameters for protein analysis with support for data warehousing and data mining. Its foundation consists of the five building blocks of data management technology: a) Data profiling: inspect data for errors, inconsistencies, redundancies, and incomplete information; b) Data quality: correct, standardize and verify data; c) Data integration: match, merge or link data from a variety of disparate sources; d) Data augmentation: enhance data using information from internal and external data sources, and e) Data monitoring: check and control data integrity over time. Currently, Sting database has over 300 parameters compiled at a single site and was implemented using MySQL1, an open-source relational database management system that uses structured query language (SQL). This database is connected to Sting, a Web-based suite of programs for comprehensive and simultaneous analysis of structure and sequence (Neshich et al., 2003, 2004a,b, 2005a,b, 2006). 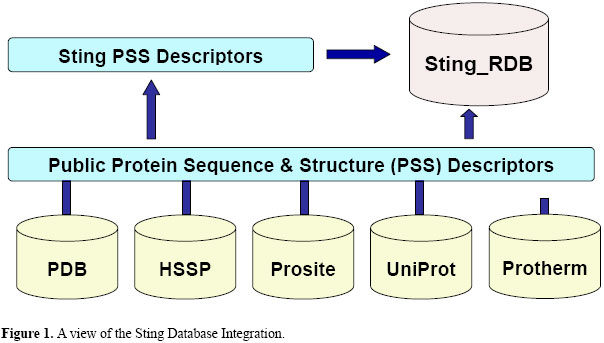 This paper differs from our previous study (Neshich et al., 2005a) in many aspects. First, we introduce Sting_RDB, a relational database with mechanisms for efficient and relevant queries using SQL. Our previous paper described a text database in which data consistency and integrity was not guaranteed. Second, we provide support for data warehousing and mining. Third, we introduce quality assessment indicator for data. Finally and probably most importantly, some complex queries that can be posed on Sting_RDB cannot be posed on text databases. STING DATABASE INTEGRATION The Sting database operates with a collection of both publicly available data (e.g., PDB (Berman et al., 2000), HSSP (Schneider and Sander, 1996; Schneider et al., 1997), Prosite (Hulo et al., 2006), and UniProt (Apweiler et al., 2004)) and its proprietary protein sequence and structure (PSS) descriptors, such as geometric parameters (e.g., cavity, curvature), physical-chemical parameters (e.g., electrostatic potential), and conservation-related parameters (e.g., SH2Qs (Higa et al., 2006), evolutionary pressure) as can be seen in Figure 1. The data consolidated and integrated into Sting_RDB make this database one of the most comprehensive databases available for analysis of protein structure and sequence. The Sting database integration is basically composed of two layers: public PSS descriptors and Sting PSS descriptors. The first layer consolidates the data available at the public databases: PDB, HSSP, Prosite, UniProt, and Protherm while the second layer computes the Sting PSS descriptors taking into account some public database parameters. Sting_RDB is updated on a weekly basis and starts by downloading the public database records. Subsequently, a script is initiated activating a set of programs designed to calculate 310 Sting PSS descriptors including geometric parameters and physical-chemical and conservation-related parameters. Finally, the most important step is performed, i.e., the Sting PSS descriptors and the public PSS descriptors are transferred from flat files to compose Sting_RDB. To accomplish that, two more scripts are used. The first one is run to normalize the PSS descriptors into 57 relational tables. This normalization refers to the process that eliminates redundancy, organizes data efficiently, and reduces the potential for anomalies during data operations and improves data consistency. The last script is run to transfer the normalized data to Sting_RDB. It is important to point out here that the five building blocks of data management (Data profiling, Data quality, Data integration, Data augmentation, and Data monitoring), mentioned in the Introduction, are taken into account during the normalization process. To accomplish that, data curation routines are used to fill in missing values, smooth noisy data and correct data inconsistencies. Consequently, the normalized database has a design that reflects the true dependencies between tracked quantities, allowing quick updates to data with little risk of introducing inconsistencies. Instead of attempting to store all information into one table, data are spread out logically into many tables. This database schema is described in the next section. STING DATABASE MODEL The relational model is a collection of one or more relations, where each relation is a table with rows and columns (Ramakrishnan and Gehrke, 2004). This simple tabular representation enables even novice users to understand the contents of a database, and it permits the use of simple, high-level languages to query the data. The major advantages of the relation model over previous data models (e.g., flat files) are its simple data representation and the ease with which even complex queries can be expressed. The Sting database model is composed of about sixty tables containing data of protein structures. Figure 2 shows a partial view of this model encompassing the major tables and their relationships. For simplicity, we only present the attributes composing the relation key, i.e., the set of attributes that uniquely identifies a record (tuple) according to a key constraint. The main construct for representing data in the relational model is a relation. A relation consists of two parts: a relation schema and a relation instance. The schema specifies the relation’s name, the name of each attribute, and the domain of each attribute. A domain is referred to in a relation schema by the domain name and has a set of associated values.
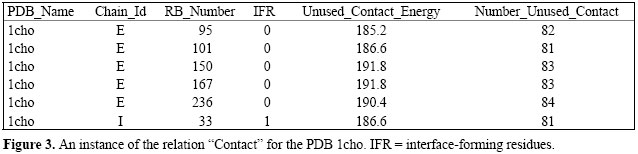
An instance of a relation is a set of tuples, also called records, in which each tuple has the same number of attributes as the relation schema. A relation instance can be thought of as a table in which each tuple is a row, and all the rows have the same number of attributes. Figure 3 illustrates these concepts for the PDB 1cho. The attribute names PDB_Name, Chain_Id, RB_Number (Residue or Base Number), IFR (interface-forming residues), Unused_Contact_Energy (expressed in kcal/Mol), Number_Unused_Contact represent the relation schema, while the rows of the tables correspond to the relation instance.
QUERYNG STING DATABASE A relational database query is a question about the data, and the answer consists of a new relation containing the result. For example, we may want to find the unused contact energy ranging from 185 to 193 kcal/Mol, as depicted in Figure 3. To accomplish that, we use a specialized language for writing queries called SQL. SQL is the most popular query language for a relational database. Figure 4 shows the SQL query related to the results retrieved in Figure 3, an example that illustrates how easily Sting_RDB can be queried.
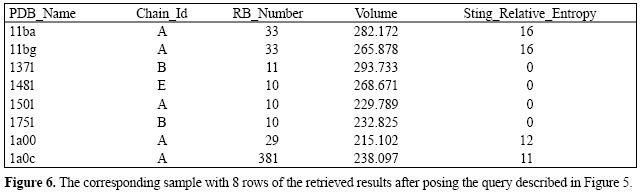
In order to show a more challenging SQL query, let us suppose we are interested in finding all the PDB files containing pockets with a volume ranging from 200 to 300 cubic angstroms and relative entropy of amino acids forming such a pocket should be less than 20. Also, the pocket-forming amino acid ensemble has to have at least one histidine (HIS) and one tyrosine (TYR). Such a query is formalized in Figure 5. The corresponding sample with the selected 8 rows from the total retrieved results can be seen in Figure 6. Of course, the relation in Figure 6 is only a sample of the 4596 results available of 37,269 PDB files. The biological meaning of this result is that all structures found could bind a metal ion, for example. Indeed, all structures listed do bind metal ions.
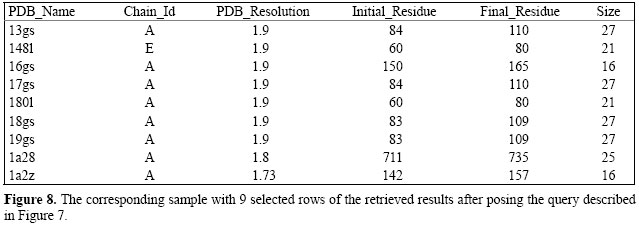
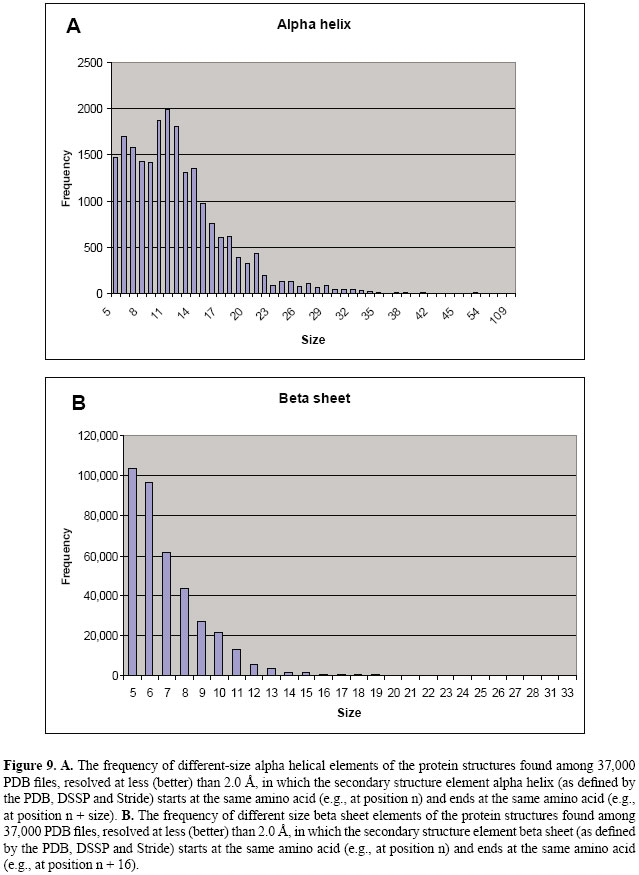
The SQL query examples 1 and 2 show how one could search the Sting_RDB for regular queries. In the next section, we show how complex queries can be supported by Sting_RDB. SUPPORT FOR DATA WAREHOUSING AND DATA MINING Some particular SQL queries may require three or more "group by", nested selects and aggregation operators such as COUNT, SUM, and AVG. These SQL queries are complex to be formalized. Apart from that, complex queries compromise the performance of a database system because these queries are time-consuming. To cope with this problem, a database administrator could design a data mart to consolidate information regarding a particular subject of the database. A data mart is a data warehouse designed primarily to address a specific function in organizations (e.g., purchasing, marketing). In other words, a data warehouse can be thought as of a collection of data marts containing information consolidated from several sources. In general, a data mart often uses aggregation or summarization of the data to enhance query performance. In particular, independent data marts are the favorite architectures for querying a data warehouse. The main reasons are that data marts simplify the data warehouse projects, reduce cost, speed up queries, and are simple to be implemented. To illustrate how Sting_RDB can be enhanced to support data warehouses, consider the following query: "Find all the alpha helices with size greater than 15 amino acids, within protein structure resolved at less (better) than 2.0 Å, in which the secondary structure element, alpha helix (as defined by the PDB, DSSP, and Stride), starts at the same amino acid (e.g., at position n) and ends at the same amino acid (e.g., at position n + 16)". Indeed, this is a very complex query and may not be answered by using a regular SQL. To deal with this particular query, we designed a data mart containing generic alpha helix information. Figure 7 shows a simpler SQL query to replace the complex query described above, while Figure 8 presents the results showing only 9 rows of 1164 retrieved results from the available 37,269 PDB files. As a consequence of being able to make queries as described in the previous two examples, we are able to maintain always updated information on, for example, size and frequency of occurrence for secondary structure elements, i.e., helix and sheet, as shown in Figure 9. When complex queries cannot be handled by designing a data mart, one could use data mining techniques to fulfill this goal. Such techniques are used for searching large volumes of data for patterns previously unknown, yielding potentially useful information (Han and Kamber, 2001). In this case, a set of programs must be used to pre-process the data before data mining takes place. One example that illustrates the potential of Sting_RDB for data mining is the method for parameter discrimination presented by Borro et al. (2006). Such a method was designed for predicting enzyme class from protein structure descriptors found in Sting_RDB using Bayesian classification.
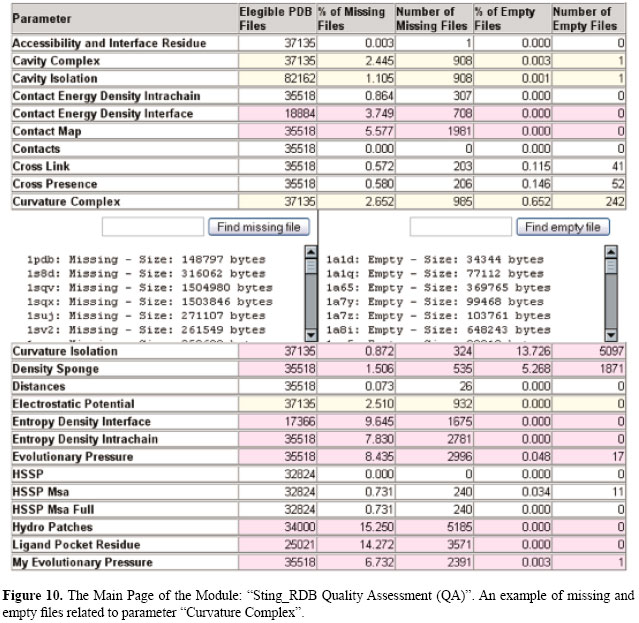
STING DATA QUALITY ASSESSMENT Considering its relevance for researchers interested in PSS analysis, the Sting_RDB has a special module to measure the quality of its data. On a weekly basis, a checklist procedure is performed to identify the parameters/files that are both missing and/or empty for the new PDB files added to the database. The main goal of such a procedure is to guarantee that the quality of the data will not be degraded as the updates take place. When the checklist procedure identifies a group of parameters that are missing and/or empty, a report is automatically sent to the Sting_RDB administrator who will run a set of scripts to update the parameters concerning the new PDB files, and subsequently, perform the checklist procedure to evaluate the quality of the updated data. In Figure 10, we show an example of missing and empty files related to the parameter "Curvature Complex". When a Sting user selects a PDB file for analysis, if one or more parameters of that PDB are not available in Sting_RDB, the user can search for such a PDB name in the Sting_RDB QA to first verify the existence of those parameters. For each parameter, there is a list of missing and empty PDB files containing the structure-related parameters. However, the situation where the data are missing is not all that frequent since we are trying to improve the quality of our DB by keeping the percentage of missing and empty structure parameters below 3% for almost all PDB files. In addition, we are working diligently to reduce that percentage to 1% or less. The main cause for the existence of such missing data is the inconsistency in PDB format files. When processed in high throughput fashion, some algorithms (responsible for the calculation of a certain parameter) will be therefore generating incomplete runs. Consequently, there is a limit imposed on us with respect to how much we could possibly improve the quality of Sting_RDB. FUTURE DEVELOPMENTS To illustrate the main features of Sting_RDB, we have created a demo which is accessible at http://www.cbi.cnptia.embrapa.br/StingRDB. A user can now exploit biologically relevant questions retrieving data and exploring relationships among them from Sting_RDB and from some data marts (e.g., alpha helix, beta sheet, and rare rotamers) derived from Sting_RDB. The demo also contains information regarding the Sting_RDB structure, its relations, and several examples of queries. However, users can also pose other queries not available as examples, in order to profit from the Sting_RDB features and content. We are diligently improving Sting_RDB in terms of content, data quality and data marts to support relevant queries that can help biologists and researchers in finding out interesting relations concerning protein sequence and structure.
CONCLUSIONS The Sting database is a relational database composed of structural parameters for protein analysis operating with a collection of both publicly available data (e.g., PDB, HSSP, Prosite) and its own data (contacts, interface contacts, surface accessibility). Currently, Sting_RDB has over 300 parameters compiled at the single site and was implemented using free software. The main features of Sting_RDB can be summarized as follows:
Apart from the features mentioned above, Sting_RDB is now going to be more accessible and readily addressable for data warehousing and mining. Most importantly, some effort has been made to make the Sting_RDB unique in terms of quality assessment when compared to other counterparts in the bioinformatics domain. To the best of our knowledge, Sting_RDB is one of the most comprehensive data repositories for PSS analysis, capable of providing its users with a data quality indicator. More importantly, biologically relevant questions can be now easily addressed by using the Sting_RDB features. REFERENCES Apweiler R, Bairoch A, Wu CH, Barker WC, et al. (2004). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32: D115-D119. Berman HM, Westbrook J, Feng Z, Gilliland G, et al. (2000). The protein data bank. Nucleic Acids Res. 28: 235-242. Borro LC, Oliveira SR, Yamagishi ME, Mancini AL, et al. (2006). Predicting enzyme class from protein structure using Bayesian classification. Genet. Mol. Res. 5: 193-202. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, et al. (2002). Intrinsic disorder and protein function. Biochemistry 41: 6573-6582. Han J and Kamber M (2001). Data mining: concepts and techniques. Morgan Kaufmann, San Francisco. Higa RH, Cruz SA, Kuser PR, Yamagishi ME, et al. (2006). Building multiple sequence alignments with a flavor of HSSP alignments. Genet. Mol. Res. 5: 127-137. Hulo N, Bairoch A, Bulliard V, Cerutti L, et al. (2006). The PROSITE database. Nucleic Acids Res. 34: D227-D230. Neshich G, Togawa RC, Mancini AL, Kuser PR, et al. (2003). STING Millennium: a web-based suite of programs for comprehensive and simultaneous analysis of protein structure and sequence. Nucleic Acids Res. 31: 3386-3392. Neshich G, Mancini AL, Kuser PR, Fileto R, et al. (2004a). Gold STING: studying protein stability and folding by looking at an extensive DB of the structure descriptors. ISMB, Glasgow. Neshich G, Rocchia W, Mancini AL, Yamagishi ME, et al. (2004b). JavaProtein Dossier: a novel web-based data visualization tool for comprehensive analysis of protein structure. Nucleic Acids Res. 32: W595-W601. Neshich G, Borro LC, Higa RH, Kuser PR, et al. (2005a). The Diamond STING server. Nucleic Acids Res. 33: W29-W35. Neshich G, Mancini AL, Yamagishi ME, Kuser PR, et al. (2005b). STING Report: convenient web-based application for graphic and tabular presentations of protein sequence, structure and function descriptors from the STING database. Nucleic Acids Res. 33: D269-D274. Neshich G, Mazoni I, Oliveira SR, Yamagishi ME, et al. (2006). The Star STING server: a multiplatform environment for protein structure analysis. Genet. Mol. Res. 5: 717-722. Qian N and Sejnowski TJ (1988). Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol. 202: 865-884. Radivojac P, Chawla NV, Dunker AK and Obradovic Z (2004). Classification and knowledge discovery in protein databases. J. Biomed. Inform. 37: 224-239. Ramakrishnan R and Gehrke J (2004). Database management systems. 3rd edn. McGraw-Hill Companies Inc., New York. Schneider R and Sander C (1996). The HSSP database of protein structure-sequence alignments. Nucleic Acids Res. 24: 201-205. Schneider R, de Daruvar A and Sander C (1997). The HSSP database of protein structure-sequence alignments. Nucleic Acids Res. 25: 226-230. 1 http://www.mysql.com/
|
|