![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Proteinase inhibition using small Bowman-Birk-type structures
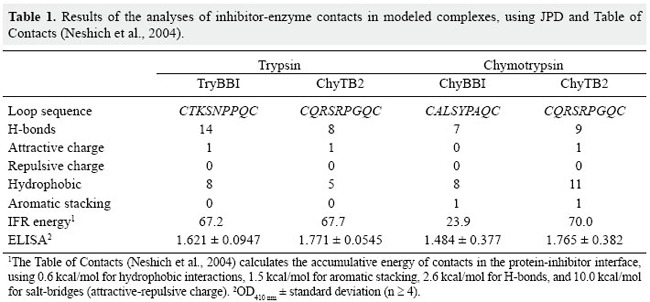
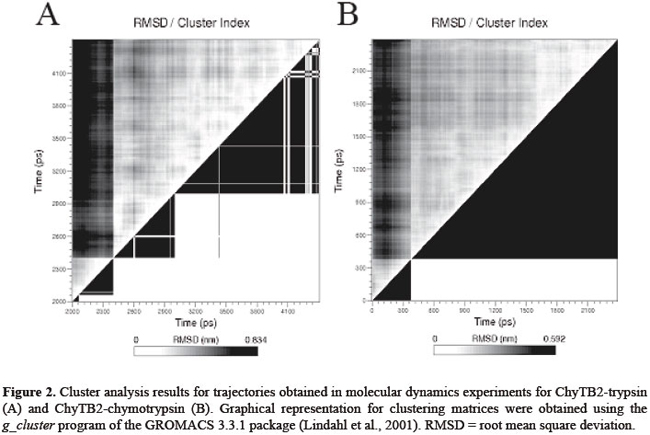
J.H. Fernandez1*, M.O. Mello2*, L. Galgaro2, A.S. Tanaka3,M.C. Silva-Filho2 and G. Neshich4 Genet. Mol. Res. 6 (4): 846-858 (2007) ABSTRACT. Bowman-Birk inhibitors (BBIs) are cysteine-rich and highly cross-linked small proteins that function as specific pseudosubstrates for digestive proteinases. They typically display a "double-headed" structure containing an independent proteinase-binding loop that can bind and inhibit trypsin, chymotrypsin and elastase. In the present study, we used computational biology to study the structural characteristics and dynamics of the inhibition mechanism of the small BBI loop expressing a 35-amino acid polypeptide (ChyTB2 inhibitor) which has coding region for the mutated chymotrypsin-inhibitory site of the soybean BBI. We found that in the BBI-trypsin inhibition complex, the most important interactions are salt bridges and hydrogen bonds, whereas in the BBI-chymotrypsin inhibition complex, the most important interactions are hydrophobic. At the same time, ChyTB2 mutant structure maintained the individual functional domain structure and excellent binding/inhibiting capacities for trypsin and chymotrypsin at the same time. These results were confirmed by enzyme-linked immunosorbend assay experiments. The results showed that modeling combined with molecular dynamics is an efficient method to describe, predict and then obtain new proteinase inhibitors. For such study, however, it is necessary to start from the sequence and structure of the mutant interacting relatively strongly with both trypsin and chymotrypsin for designing the small BBI-type inhibitor against proteinases. Key words: Bowman-Birk inhibitor, Molecular modeling, Enzyme-linked immunosorbent assay, Enzyme specificity INTRODUCTION Plant defense mechanisms against herbivorous insects include the production of serine proteinase inhibitors that affect insect development (Pompermayer et al., 2001). One group of these inhibitors, called Bowman-Birk inhibitors (BBIs), function as specific pseudosubstrates for the digestive proteinases, forming a stable complex in which proteolysis is limited and extremely slow (Tiffin and Gaut, 2001). As a consequence, an amino acid deficiency occurs in insects feeding on such diet and which consequently affects the insect growth, development and fecundity (Pompermayer et al., 2001). BBIs are cysteine-rich and highly cross-linked small proteins that typically display a "double-headed" structure (Rahbe et al., 2003). Each "head" contains an independent proteinase-binding loop that can bind and inhibit trypsin, chymotrypsin and elastase (McBride et al., 2002; Singh and Appu Rao, 2002) either independently or simultaneously. The resulting non-covalent complex renders the proteinase inactive. The realization that one BBI molecule could form a 1:1:1 complex with two enzymes led early workers to dissect this activity. This inhibition mechanism is common for the majority of serine proteinase inhibitor proteins, and many analogous examples are known. A particular feature of the BBI protein is that the interacting loop is a particularly well-defined disulfide-linked short beta-sheet region. Moreover, small synthetic peptides based on this region keep the same structure as the corresponding part of the full-sized protein and also retain inhibitory activity (Brauer et al., 2002). It has been possible to isolate the antiproteinase activity as small (approximately 11 residues), cyclic, synthetic peptides, which display most of the functional aspects of the protein (Brauer et al., 2002). Based on these characteristics, some serine proteinase inhibitors were over expressed in plants aiming to increase their resistance to insects. In some cases, this expected effect was satisfactorily observed, but in others, a variable level of resistance was achieved (De Leo et al., 2001; Falco and Silva-Filho, 2003). In recent experiments with BBI-type inhibitors described by Mello (2002), a mutated sequence was obtained (for the region 31-58) for the BBI proteinase inhibitor extracted from Glycine max (P01055, 1D6R:I structure). The mutant has the inhibition loop formed by the residues CTRSIPPQC and was called ChyTB2. In the present study, we used computational biology to study the structural characteristics of ChyTB2 loop, which was described as a good inhibitor for bovine trypsin and chymotrypsin. Modeling and unrestrained molecular dynamics was used to allow the final accommodation of the modeled enzyme-inhibitor complexes. The stability, hydrogen bonds, and electrostatic and hydrophobic interactions of each complex were compared to already described BBI interactions with trypsin and chymotrypsin. In our opinion, the inhibitor studied here may be used as a lead for the development of small and effective proteinase inhibitors. MATERIAL AND METHODS Cloning of BBI derivatives The soybean BBI cDNA (GenBank access P01055) sequence was silent mutated at positions Ser38 (TCG→TCA) and Cys39 (TGT→TGC) to insert the unique restriction site NsiI. The restriction sites PstI and KpnI were inserted at the ends of the sequence to be cloned into the pCTB vector. The construction was confirmed by sequencing. Two different PCR amplifications of phagemid pCTB-BBI were used in the division of the bbi gene. The T1 (5’-ccgggctgcagaattcgagctcggt-3’) and T2 (5’-cctctgcagaatgcatgatttacaagc-3’) (both PstI tailed) primers were used to amplify the trypsin (TryBBI) portion, whereas the chymotrypsin (ChyBBI) fragment was amplified using C1 (5’-cgcggtaccctgcaggttttccttg-3’) and C2 (5’-cggtaccaaatcatgcatttgcgc-3’) primers (Operon, California, USA), both KpnI tailed. The PCR products were digested with the appropriate restriction enzyme and ligated to the pCTB vector, thus allowing for the recombinant phagemid pCTB-TryBBI and pCTB-ChyBBI. Both constructions of BBI derivatives were confirmed by sequencing and expressed on the tips of phages. The division of the former bbi sequence generated two fragments: one expressing a 42-amino acid polypeptide (hereafter referred to as TryBBI) and containing the coding region for the trypsin inhibitory site, and the second one expressing a 35-amino acid polypeptide (hereafter referred to as ChyBBI) and containing the coding region for the chymotrypsin inhibitory site. Construction of TryBBI and ChyBBI mutant libraries The phage-display libraries were constructed varying amino acids at 4 positions in TryBBI and ChyBBI. Two degenerated oligonucleotides T-NNB (5’-gatcaatgcgcatgcNNBNNBNNBNNBcctcctcaatgccgctgttca gatatgagactgaattcgtgccattcagcttgtaaatcatgcatttgcgcac-3’) and C-NNB (5’-ggtaccaaatcatgcatttgcNNBNNB tcgNNBcctNNBcagtgtttttgtgtcgacataacc gatttc-3’) were synthesized containing the nucleotide sequence NNB (B = C/G/T, N = A/C/G/T) which codes for a restricted pool of amino acids at the positions P2, P1, P1’, P2’ of TryBBI and P2, P1, P2’, P4’ of ChyBBI. A PCR amplification of these two degenerated oligonucleotides using their backward primers T-NNBback (5’-gtgcgcaaatgcatgattta-3’) and C-NNBback (5’-gaaatcggttat gtcgacac3’) was performed as described by Tanaka et al. (1999). The resulting double-stranded material was cleaved with SphI-NsiI for the trypsin inhibitors, and NsiI-SalI for the chymotrypsin inhibitors. After restriction, fragments were ligated into dephosphorylated phagemid vectors pCTB-TryBBI and pCTB-ChyBBI, respectively. A mix of ligation products was used to transform Escherichia coli TG1 cells (K12(lac-pro), supE, thi, hsdD5/F’, traD36, proAB, lacIq, lacZ∆M15), creating the trypsin and chymotrypsin inhibitor libraries. Selection of the phage-display libraries Library amplification, rescue of recombinant phage-display libraries and selection were performed as described by Mello (2002). Selection was carried out with bovine trypsin (0.2 mg/mL in 50 mM NaHCO3, pH 9.6) immobilized on immunotubes (Maxi Sorb 4070319, Nunc). After five rounds of selection, bound fusion phages were eluted and used for transfection of E. coli cells. One hundred colonies of each selection were sequenced and evaluated for further analysis. Enzyme-linked immunosorbent assay (ELISA) of phages was performed using the Detection Module-Recombinant Phage Antibody System (Pharmacia) as described in the instruction manual. Microtiter plates (Costar) were coated with bovine trypsin and chymotrypsin (Sigma), papain (Sigma, negative control) and sugar cane borer trypsin (1 µg/mL purified protein). Molecular modeling The structure of the cancer chemopreventive BBI complexed with bovine trypsin (pdb 1D6R) (Koepke et al., 2000) has been used to predict the 3-D structure of the TryBBI (1-36 residues of the I chain) and ChyBBI loops (31-58 residues of the I chain) of modeled inhibitors. The atomic coordinates of the 1D6R pdb complex, solved at 2.4 Å resolution, were used as a template in comparative modeling by satisfaction of spatial restraints (Fiser et al., 2000) implemented in the program Modeller 6v2 (Sali and Blundell, 1993). The structure of the bovine gamma chymotrypsin (pdb 1GMC) (Yennawar et al., 1994) was used in the modeling of the bovine chymotrypsin-ChyBBI complex. Final interaction model for the complex was obtained by satisfaction of spatial restrains (Fiser et al., 2000) in MODELLER 6v2, implementing CHARMM energy terms for five cycles of simulated annealing (Marti-Renom et al., 2000). For each complex, 25 different models were generated using MODELLER script, and the quality of the predicted folds was evaluated using the internal score of the variable target function (Marti-Renom et al., 2000). The stereochemical quality of the 3 best scoring models was assessed by Procheck program (Laskowski et al., 1993) at the same resolution as the 1D6R structure. The final model was selected based on the overall stereochemical quality for further energy minimization using Gromacs (Lindahl et al., 2001). Energy minimization and molecular dynamics of modeled complexes To refine the models of the enzyme-BBI complexes and to allow best accommodation of the contacting residues in the enzyme-inhibitor interface, additional energy minimization and equilibrating molecular dynamics simulations were carried out with the program Gromacs 3.2 (Lindahl et al., 2001) on a dual-CPU Linux work station. Complexes were solvated in explicit single point charge solvent model, using octahedral water box subjected to periodic boundary conditions and ionizable amino acids were protonated according to pH 7.0. The simulated system (~39,000 atoms) was fully solvated with 12,000 water molecules. To neutralize the system, one Na+ and 5 Cl- ions were added in the box. Initial complex models were submitted to a steepest-decent energy minimization (5000 steps), to remove bad van der Waals contacts. For further relaxation, the minimized structure of the complexes was used in unrestrained molecular dynamics for 500 ps with berendsen-type temperature (312 K) and pressure (1 atm.) coupling in an 8 x 12 x 8-nm simulation cell, implementing the smooth particle mesh Ewald method of electrostatic treatment (Essman et al., 1995). Production dynamics were carried out under the same conditions for 2-3 ns. Cluster analysis of the trajectory obtained was performed using g_cluster program of Gromacs 3.2 (Lindahl et al., 2001) with 2.5 Å cut-off to obtain the average structures of the complex during production simulation. The most stable complex in the MD run was selected for unrestrained multiple step conjugate-gradient minimization process (0.1 kJ mol-1 nm-1) to obtain the final minimized structure of the complex for further analysis. Evaluation of protein-inhibitor interactions in enzyme-BBI complexes For evaluation of the trypsin-inhibitor and chymotrypsin-inhibitor interactions, the Java Protein Dossier (JPD, Neshich et al., 2004) program of the Diamond Sting Suite (Neshich et al., 2005) was used. The cut-off distances for hydrogen bonds, salt bridges, and aromatic interactions were 2-3.2, 2-6 and 4.0 Å, respectively. To evaluate the stability of the secondary structure elements at the contact surface of the enzyme-inhibitor complex, the program do_dssp in Gromacs package (Lindahl et al., 2001) was used. RESULTS Structure of the TryBBI and ChyBBI models In order to study the TryBBI and ChyBBI interactions, the sequence of soybean BBI was used to model and analyze the inhibition mechanism for each complex. The 1-36 residues in the sequence/structure of soybean BBI (pdb 1D6R:I) (Lindahl et al., 2001) were used to model our TryBBI, and 31-58 residues of the same sequence/structure were used to model the ChyBBI. After the modeling procedure and validation of the model using Procheck, molecular dynamics simulation was used to allow the proper accommodation of new "in silico" constructed complexes (Figure 1A and B). A 15-residue finger-like structure, but with different physicochemical characteristics composes each BBI loop. The TryBBI-interacting loop (Figure 1A) is composed mainly of charged and polar residues. At the same time, the ChyBBI-interacting loop (Figure 1B) is composed of hydrophobic residues. Contact surface differences in bovine TryBBI- and ChyBBI-modeled complexes In BBI-trypsin complex (1D6R pdb), the important BBI-contacting residues are Lys16 and Asn18 at the "left" side, Gln21 at the "right" side and Arg23 at the "upper" side of the contacting surface (Figure 1A). The most important residue in the contact interface is Lys16, able to make a salt bridge with Asp189, and hydrogen bonds with Gly193 and Ser214. The Cys14-Gly216 hydrogen bond at the center of the β-barrel structure of the BBI loop, and Arg23-Asn97 and Cys12-Ser217 hydrogen bonds stabilize the position of the BBI at the upper and right sides of the contact interface. 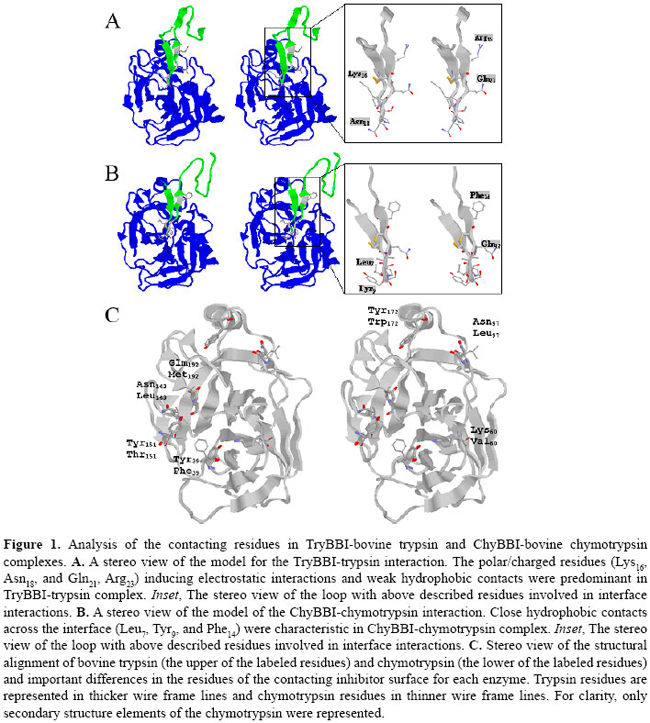 In our modeled BBI-chymotrypsin complex, the most important BBI-contacting residues are Leu7 and Tyr9 at the "left" side, Gln12 at the "right" side and Phe14 at the "upper" side of the contacting surface (Figure 1B). The most important residue in the contact interface is Leu7, able to make hydrogen bonds with Gly174 and Ser195, and hydrophobic contact with Leu180 and Ala194 ring. The hydrophobic pocket formed by Trp153, Trp196 and Ile82 residues of chymotrypsin allows the accommodation of the Ile4 and Phe14 BBI residues at the upper side of the contact surface through hydrophobic interactions. The position of the Tyr9 is coordinated by aromatic stacking with Phe22 and a hydrogen bond with Phe24 enzyme residues. The Cys5-Gly197 and Ser8-His40 hydrogen bonds also maintain the overall secondary structure of the ChyBBI loop-chymotrypsin complex. After structural alignment in JPD, bovine trypsin (pdb 1D6R) (Koepke et al., 2000) and chymotrypsin (pdb 1GMC) (Yennawar et al., 1994) were fitted at less than 1.5 Å RMSD and residues located on the inhibitor-contacting surface were compared. The important residues in the inhibitor-contacting surface were positioned in a ring form, comprising the 97 and 172 positions at the "upper" side, 151, 143 and 192 positions at the "left" side, and 39 and 60 positions at the "right" side of the surface (Figure 1C). The Asn-Leu change at 97 and 143 positions, and the Tyr-Phe39, Lys-Val60, Asn-Leu143, Tyr-Trp172, and Gln-Met192 substitutions were mapped in the trypsin/chymotrypsin structures, and are shown in Figure 1C. The physicochemical properties in these 7 different residues are responsible for the general change of polar to hydrophobic contacting at interface in the trypsin and chymotrypsin enzymes, respectively. Structural model for the ChyTB2 inhibition mechanism Obtained in phage-display experiments, the ChyTB2 mutant (CTRSIPPQC loop) showed high affinity for bovine trypsin and chymotrypsin enzymes in ELISA experiments (Table 1). These characteristics are ideal for developing a good inhibitor for both enzymes. ChyTB2 inhibitor in complex with bovine trypsin (ChyTB2-Try) and bovine chymotrypsin (ChyTB2-Chy) were modeled and submitted to molecular dynamics simulation in order to study the dynamics of inhibition and the important residues to both these complexes. The molecular dynamics trajectories obtained were submitted to cluster analysis (Figure 2), and the representative structure of the most represented cluster was used as the complex structure for interaction analysis. For ChyTB2-Try complex, a stable cluster was obtained in 3.8-4.2 ns of dynamics (Figure 2A), and the representative structure for further analysis was tacked for 3800 ps. For ChyTB2-Chy complex, stable complex was obtained after 700 ps of dynamics (Figure 2B), and representative structure was tacked for 1800 ps.
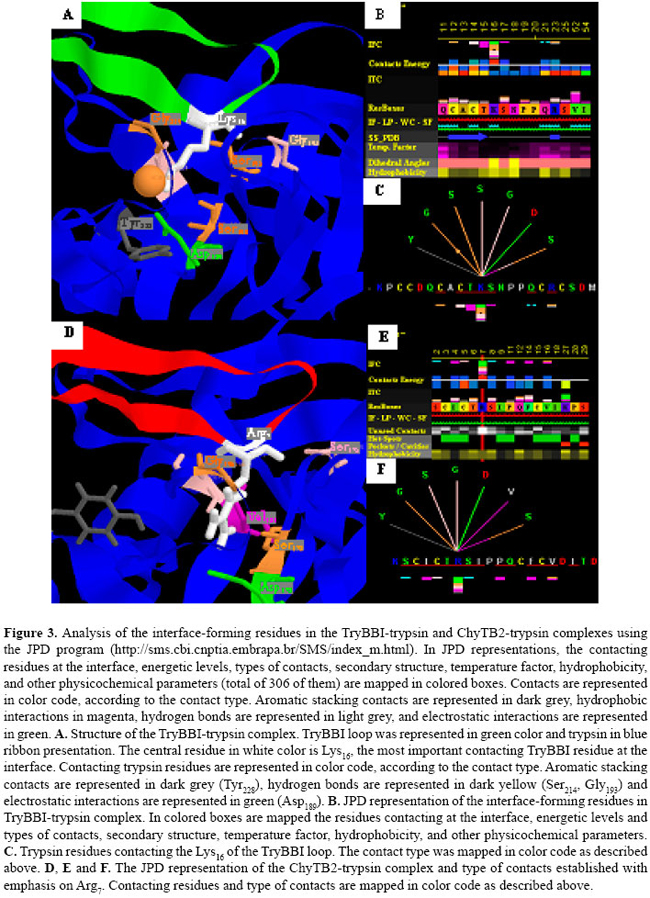
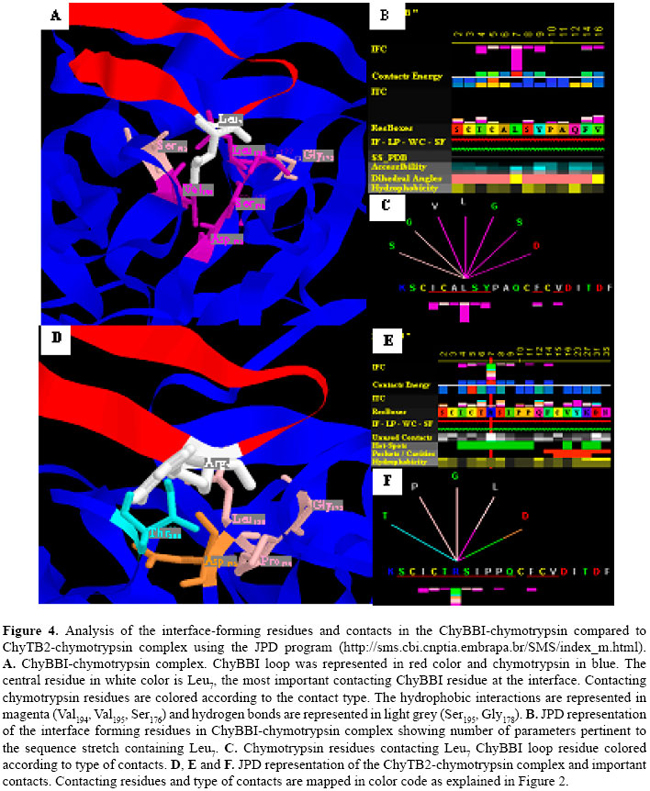
Although the ChyTB2 mutant maintains the hydrophobic characteristics of the ChyBBI type loop, an Arg present at position P1 allows for the formation of salt bridges and hydrogen bonds with residues of the bovine trypsin (Figures 3 and 4). As we described above, in the TryBBI-trypsin complex the most important interactions are salt bridges and hydrogen bonds, and the central residue in the interface is the Lys16 at the P1 loop position. In our "in silico" experiments, the Lys16 residue at P1 loop position is able to make a salt bridge with Asp189, and hydrogen bonds with Gly193 and Ser214 enzyme residues (Figure 3A). In the ChyTB2 mutant, the Lys residue is replaced by Arg at the P1 loop position, allowing the formation of a salt bridge with Asp189 and hydrogen bond with Gly193 and Ser214 (Figure 3A-C). In the direct comparison, although the trypsin contacting residues in TryBBI and ChyTB2 inhibitors are different (Figure 3B,E), the Lys16-contacting residues in the TryBBI-trypsin complex, Arg16-contacting residues in ChyTB2-trypsin complexes, and the type of contacts in both complexes are almost the same (Figure 3C,F). At the same time, our "in silico" experiments showed that in the ChyBBI-chymotrypsin complex, the most important interactions are hydrophobic. The Leu7 residue (P1 loop position) is anchored through hydrophobic interactions and makes hydrogen bonds with Gly174 and Ser195 in the ChyBBI-chymotrypsin complex (Figure 4A-C). The ChyTB2 mutant interacts with the bovine chymotrypsin hydrophobic surface through Ile, Thr, Ile, Pro, and Phe residues. The Arg7 residue at the P1 loop position maintains a hydrogen bond with the Gly178, making a new one with Pro179 and Leu180, and a salt bridge with Asp175 (Figure 4D-F).
As the ChyTB2 mutant in ELISA experiments revealed significantly enhanced binding capacity with trypsin and chymotrypsin as compared to the native BBI (Table 1), the sequence of this mutant must be used as a starting structure for a BBI-type inhibitor for both enzymes DISCUSSION The ChyTB2 (CTRSIPPQC) mutant’s high affinity for both bovine trypsin and chymotrypsin in ELISA experiments (Table 1) is an ideal characteristic to maintain in the development of a strong dual enzyme inhibitor. Although this mutant maintains the hydrophobic characteristics of the ChyBBI-type loop and continues to interact with the bovine chymotrypsin hydrophobic surface through Thr, Ile and Pro residues at the P2, P2’ and P4’ sites, respectively (Figure 4), it also has an Arg at position P1 that allows the formation of salt bridges with Asn189 and hydrogen bonds with the Gly193 and Ser214 residues of bovine trypsin (Figure 3). An interesting observation is that the sequence of the inhibitory loop in this molecule occurs naturally in five different BBIs from pea (CAC24566), alfalfa (S56647, P80321, P16346) and peanut (P01067) (Mello et al., 2003). Moreover, one of the alfalfa inhibitors (S56647) is a double-headed BBI that contains the same sequence in both heads. Single-headed inhibitors of serine proteinases, which can interact with both trypsin and chymotrypsin, have been reported in potato (Pearce et al., 1982; Moura and Ryan, 2001), and more effective inhibition of chymotrypsin by a trypsin inhibitor was also observed by Moura and Ryan (2001) in pepper. The P1 position of proteinase inhibitors determines up to 40-70% of the total association energy of the complex formation (Lu et al., 1997). Positively charged Arg residues are able to make hydrogen bonds, salt bridges and even aromatic stacking type contacts (Figures 3 and 4). Another position that plays an important role is P2’ since it fits into an apolar S2 enzyme pocket. P2’ Ile and Met are the residues most frequently found in BBIs from dicotyledonous plants, present in 78 of 121 inhibitory loops from 61 different dicotyledonous species. Gariani and co-workers (1999) studying the residues at the P2’ position, observed that large aliphatic side chains give the best inhibitors while positively charged residues are tolerated at this position and negatively charged side chains give poor inhibitors. According to Brauer and Leatherbarrow (2003), when Pro is present at the P4’ position, an additional restraint on the backbone of the peptide occurs which appears to reduce the rate of tryptic hydrolysis. The results showed that modeling combined with molecular dynamics is an efficient method to obtain new proteinase inhibitors. Therefore, the sequence of the mutant strongly interacting with both types of enzymes (trypsin and chymotrypsin) should be used as a starting structure for constructing an efficient BBI-type inhibitor for both chymotrypsin- and trypsin-type enzymes. Our contribution also emphasizes the need for having an integrated environment that can display the highest possible number of parameters, which could influence the specificity of binding. Such environment has been proven to add to both the efficiency of the work and more complete analysis of the system. The STING environment in its current Diamond version (Neshich et al., 2005) has been very helpful in order to acquire and simultaneously display and analyze the largest number of sequence/structure/function attributes relevant for the current study. ACKNOWLEDGMENTS Research supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) through grants 97/04934-3 and 03/00785-6, CNPq 521093/2001-5 (NV). Special acknowledgments go to Neusa Lima and Patricia E.F. Moraes for secretarial assistance. REFERENCES Brauer AB and Leatherbarrow RJ (2003). The conserved P1’ Ser of Bowman-Birk-type proteinase inhibitors is not essential for the integrity of the reactive site loop. Biochem. Biophys. Res. Commun. 308: 300-305. Brauer AB, Kelly G, Matthews SJ and Leatherbarrow RJ (2002). The (1)H-NMR solution structure of the antitryptic core peptide of Bowman-Birk inhibitor proteins: a minimal canonical loop. J. Biomol. Struct. Dyn. 20: 59-70. De Leo F, Bonadé-Bottino M, Ceci LR, Gallerani R, et al. (2001). Effects of a mustard trypsin inhibitor expressed in different plants on three lepidopteran pests. Insect Biochem. Mol. Biol. 31: 593-602. Essman U, Perela L, Berkowitz ML, Darden T, et al. (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103: 8577-8592. Falco MC and Silva-Filho MC (2003). Expression of soybean proteinase inhibitors in transgenic sugarcane plants: effects on natural defense against Diatraea saccharalis. Plant Physiol. Biochem. 41: 761-766. Fiser A, Do RK and Sali A (2000). Modeling of loops in protein structures. Protein Sci. 9: 1753-1773. Gariani T, McBride JD and Leatherbarrow RJ (1999). The role of the P2’ position of Bowman-Birk proteinase inhibitor in the inhibition of trypsin. Studies on P2’ variation in cyclic peptides encompassing the reactive site loop. Biochim. Biophys. Acta 1431: 232-237. Koepke J, Ermler U, Warkentin E, Wenzl G, et al. (2000). Crystal structure of cancer chemopreventive Bowman-Birk inhibitor in ternary complex with bovine trypsin at 2.3 Å resolution. Structural basis of Janus-faced serine protease inhibitor specificity. J. Mol. Biol. 298: 477-491. Laskowski RA, MacArthur MW, Moss DS and Thornton JM (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26: 283-291. Lindahl E, Hess B and van der Spoel D (2001). GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 7: 306-317. Lu W, Apostol I, Qasim MA, Warne N, et al. (1997). Binding of amino acid side-chains to S1 cavities of serine proteinases. J. Mol. Biol. 266: 441-461. Marti-Renom MA, Stuart AC, Fiser A, Sánchez R, et al. (2000). Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29: 291-325. McBride JD, Watson EM, Brauer AB, Jaulent AM, et al. (2002). Peptide mimics of the Bowman-Birk inhibitor reactive site loop. Biopolymers 66: 79-92. Mello MO (2002). Inibidores de proteinase do tipo Bowman-Birk: evolução molecular, expressão na superfície de fagos filamentosos e seu papel na interação planta-inseto. PhD thesis, University of São Paulo, São Paulo. Mello MO, Tanaka AS and Silva-Filho MC (2003). Molecular evolution of Bowman-Birk type proteinase inhibitors in flowering plants. Mol. Phylogenet. Evol. 27: 103-112. Moura DS and Ryan CA (2001). Wound-inducible proteinase inhibitors in pepper. Differential regulation upon wounding, systemin, and methyl jasmonate. Plant Physiol. 126: 289-298. Neshich G, Rocchia W, Mancini AL, Yamagishi ME, et al. (2004). JavaProtein Dossier: a novel web-based data visualization tool for comprehensive analysis of protein structure. Nucleic Acids Res. 32: W595-W601. Neshich G, Borro LC, Higa RH, Kuser PR, et al. (2005). The Diamond STING server. Nucleic Acids Res. 33: W29-W35. Pearce G, Sy L, Russell C, Ryan CA, et al. (1982). Isolation and characterization from potato tubers of two polypeptide inhibitors of serine proteinases. Arch. Biochem. Biophys. 213: 456-462. Pompermayer P, Lopes AR, Terra WR, Parra JRP, et al. (2001). Effects of the soybean proteinase inhibitors on development, survival and reproductive potential of the sugarcane borer, Diatraea saccharalis. Entomol. Exp. Appl. 99: 79-85. Rahbe Y, Ferrasson E, Rabesona H and Quillien L (2003). Toxicity to the pea aphid Acyrthosiphon pisum of antichymotrypsin Sali A and Blundell TL (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234: 779-815. Singh RR and Appu Rao AG (2002). Reductive unfolding and oxidative refolding of a Bowman-Birk inhibitor from horsegram seeds (Dolichos biflorus): evidence for “hyperreactive” disulfide bonds and rate-limiting nature of disulfide isomerization in folding. Biochim. Biophys. Acta 1597: 280-291. Tiffin P and Gaut BS (2001). Molecular evolution of the wound-induced serine protease inhibitor wip1 in Zea and related genera. Mol. Biol. Evol. 18: 2092-2101. Yennawar NH, Yennawar HP and Farber GK (1994). X-ray crystal structure of gamma-chymotrypsin in hexane. Biochemistry 33: 7326-7336. |
|