![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
Intraflagellar transport complex in Leishmania spp. In silico genome-wide screening and annotation of gene function J.J.S. Gouveia, E.J.R. Vasconcelos, A.C.L. Pacheco, R. Araújo-Filho, A.R.S. Maia, M.T. Kamimura, M.P. Costa, D.A. Viana, R.B. Costa, R. Maggioni and D.M. Oliveira Genet. Mol. Res. 6 (4): 766-798 (2007) ABSTRACT. Flagella are constructed and maintained through the highly conserved process of intraflagellar transport (IFT), which is a rapid movement of particles along the axonemal microtubules of cilia/flagella. Particles that are transported by IFT are composed of several protein subunits comprising two complexes (A and B), which are conserved among green algae, nematodes, and vertebrates. To determine whether or not homologues to members of the IFT complex proteins are conserved in Leishmania spp, we scanned genomes, transcriptomes and proteomes of Leishmania species in a search for putative IFT factors, which were then identified in silico, compared, cataloged, and characterized. Since a large proportion of newly identified genes in L. major remain unclassified, with many of these being potentially Leishmania- (or kinetoplastid-) specific, there is a need for detailed analyses of homologs/orthologs that could help us understand the functional assignment of these gene products. We used a combination of integrated bioinformatics tools in a pathogenomics approach to contribute to the annotation of Leishmania genomes, particularly regarding flagellar genes and their roles in pathogenesis. This resulted in the formal in silico identification of eight of these homologs in Leishmania (IFT subunits, 20, 27, 46, 52, 57, 88, 140, and 172), along with others (IFTs 71, 74/72, and 81), as well as sequence comparisons and structural predictions. IFT, an important flagellar pathway in Leishmania, begins to be revealed through screening of trypanosomatid genomes; this information could also be used to better understand fundamental processes in Leishmania, such as motility and pathogenesis. Key words: Intraflagellar transport, Leishmania, Flagellar motility, Intraflagellar transport complex, Genome screens, Bioinformatics INTRODUCTION Leishmania protozoa are responsible for a group of diseases, collectively known as Leishmaniasis. They are trypanosomatid members of the order Kinetoplastida, which contains other important uniflagellate pathogens, such as Trypanosoma cruzi and T. brucei. Studies across eukaryotic systems indicate that flagella are constructed and maintained through the highly conserved process of intraflagellar transport (IFT), for which many of the proteins involved have yet to be identified (Haycraft et al., 2003). Well characterized in the biflagellate Chlamydomonas, IFT is a rapid movement of particles along the axonemal microtubules of cilia and flagella (Rosenbaum et al., 1999); it is a specialized bidirectional transport process mediated by the ancestral and conserved IFT complex. Import and export of proteins appear to be largely mediated by IFT particles that move along the axonemal doublet microtubules, just beneath the flagellar membrane (Kozminski et al., 1995; Rosenbaum and Witman, 2002; Cole, 2003); these are associated with either kinesin or dynein motor proteins, recycling kinesin and discarding axoneme proteins back to the cytosol (Tull et al., 2004). The main function of IFT is likely to be the delivery of axonemal substructures from the basal body region to the distal end of the flagellum, where the axoneme is assembled (Johnson and Rosenbaum, 1992; Piperno et al., 1996; Iomini et al., 2001). The particles that are transported by IFT are composed of several protein subunits (Piperno and Mead, 1997; Cole et al., 1998). The functions of the individual subunits are not known, but the proteins are conserved among green algae, nematodes, and vertebrates (Cole et al., 1998; Rosenbaum et al., 1999). In Chlamydomonas, the IFT particles comprise two large complexes: complex A is composed of six subunits (IFT-122A/B, -139, -140, -144/148, and IFT43); complex B is composed of 11 subunits (IFT20, 27, 46, 52, 57, 74/72, 80, 81, 88, and 172) (Cole et al., 1998). Morphologically similar particles were observed in trypanosomatids (Sherwin and Gull, 1989), but it is unknown whether they represent the functional equivalent of the subunits described in Chlamydomonas (Ersfeld and Gull, 2000). The recent development of robust molecular genetic and proteomic approaches (Acestor et al., 2002; El Fakhry et al., 2002; Drummelsmith et al., 2003, 2004), coupled with ongoing analysis of the genome sequences of L. major, L. infantum and L. chagasi, as well as the available related genomes of T. cruzi, T. brucei and T. gambiensi, provided us with plenty of data to apply computational biology tools in order to improve the search for and the analyses of putative IFT factors in genomes, transcriptomes and proteomes of Leishmania species. Our goal was to help annotate of Leishmania gene products, particularly those involved in the flagellar apparatus and its role in pathogenesis. Genetic studies in Chlamydomonas have demonstrated that the motility of IFT particles or individual IFT components requires the activity of kinesin-2 for anterograde movement and cytoplasmic dynein 1b for retrograde movement (Cole, 2003). Previously we reported potential virulence factors of Leishmania spp that are also components of the flagellar structure, or that are directly related to it (Oliveira et al., 2005); more recently (Vasconcelos et al., 2007) we outlined some IFT-related factors (such as profilins, katanins and kinesin homologues) that were assessed in silico and selected for possible roles in flagellar assembly, disassembly and dynamics. Here, we have begun to explore flagellar proteins that are conserved as specific components of the IFT complex in trypanosomatids. MATERIAL AND METHODS Biological databases and bioinformatics tools A complete list of data sources and tool references (we used publicly available datasets of individual or clusters of gene/protein data on Leishmania spp, mainly L. major and L. infantum) are depicted in Table 1, which includes whole genome shotgun (WGS) strategy projects, and cellular and flagellar proteome analyses of Leishmania and related eukaryotic organisms, including C. reinhardtii, for specific flagellar genes. For database searches (Table 1), as previously described (Oliveira et al., 2005), programs such as variants of BLAST (Altschul et al., 1997) and GlimmerHMM (Majoros et al., 2004) have been widely used for sequence similarity searches, comparisons and gene predictions; the resulting data were built into a local dataset that has evolved to be an organellar database (named FlagelLink - http://nugen.uece.br/flagellink/; Araújo FF, Alcoforado WJO, Lira JD, Tavares DB, et al., personal communication) suitable for subsequent searches. External database search results for WGS/CDS and individual remote sequence matches were included in our local dataset. We used MUSCLE (Edgar, 2004) for global analysis of protein sequences through multiple alignments. In silico survey We took alignments created with FASTA/BLAST as input and computed alignment tables, providing hierarchical and successive correlations between each two sets of sequences. Given that ~70% of the genes in L. major genome have no significant similarity to existing genes in sequence databases (Aggarwal et al., 2003) and that the number of experimentally confirmed gene predictions in Leishmania was small, we had to extract a large number of consensus sequences for each Leishmania species by examining for putative protein-coding open-reading frames, combining gene prediction tools with semi-automated procedures (for detailed workflow, see Oliveira et al., 2005). FASTA files for amino-acid sequences of coding regions were downloaded from the sources detailed in Table 1. Briefly, we ran local BLASTP (Altschul et al., 1997) in order to determine the sequence similarity among all coding regions. Gene/open-reading frame identifiers were used to compare the sequence data with some available expression profiles of model organisms. Our screens included local BLASTP searches of publicly available databases (NCBI non-redundant protein database, GeneDB database and UniProt/Swiss-Prot/trEMBL knowledge database (accession numbers/identifiers are those used in these three databases), searched against various collections of protein motifs and families (listed in Table 1). Gene ontology terms were assigned, based on top matches to proteins with gene ontology annotations from Swiss-Prot/tremble (www.expasy.org/sprot), AMIGO after GeneDB (www.genedb.org/amigo/perl) and TargetP (www.cbs.dtu.dk/services/TargetP) access. The functional assignment of these genes/gene products was inferred using an RPS-BLAST search against conserved domain databases (Marchler-Bauer et al., 2005); information was taken into account about subcellular localization (Emanuelsson et al., 2000), sequence and structural features, domains/motifs conservation (von Mering et al., 2005; Letunic et al., 2006) and in vitro characterization (Avidor-Reiss et al., 2004; Tull et al., 2004; Pazour et al., 2005). Proteins annotated by Swiss-Prot as being encoded in an organelle functionally or structurally related to the flagellar membranes, or containing an organelle transit peptide according to TargetP (Emanuelsson et al., 2000), were specifically incorporated in our local database. For in silico assessment of generally stored information about interactions and reactions, as well as detailed information about molecular mechanisms and underlying ontologies, we used BIND (Biomolecular Interaction Network Database) v3.7 at http://bind.ca (Alfarano et al., 2005). 
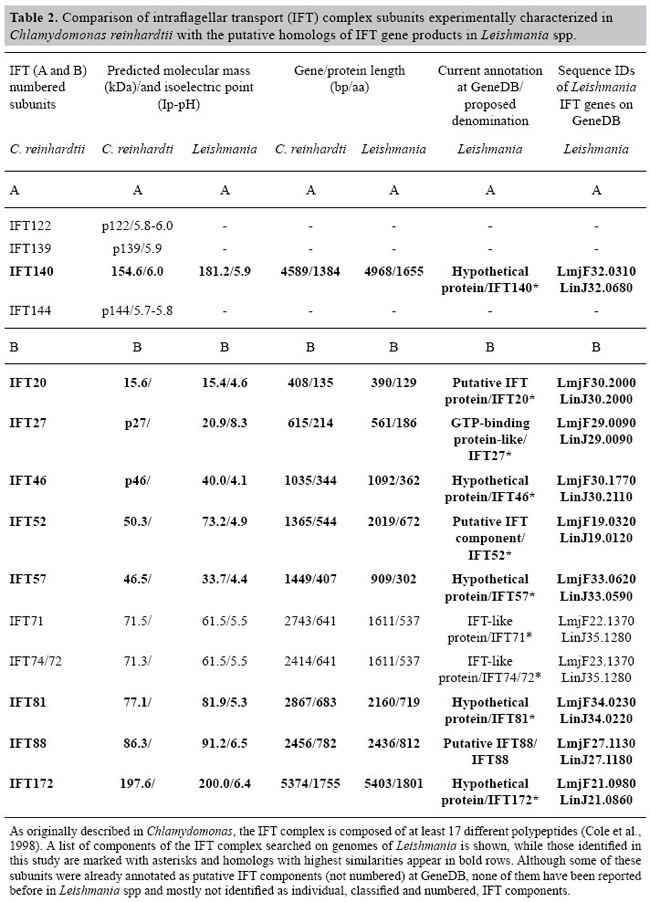
RESULTS AND DISCUSSION Leishmania genes homologous to the components of intraflagellar transport complex A and BFor the in silico identification of IFT genes/proteins in Leishmania, we adopted an approach that investigates all components of the machinery, including IFT particles and stationary elements, such as axonemes, basal bodies, and distal structures of flagella. For instance, we used models described by Iomini et al. (2001) and Parker and Quarmby (2003) for intraflagellar particle recycling. Through this approach, we were able to distinguish several Leishmania flagellar genes whose predicted proteins are either actin-, tubulin-, axoneme- or microtubule-related and that could be directly assigned as components of the IFT complex in Leishmania spp (Table 2). The functional assignment of IFT genes/gene products was inferred from local and global alignments (see Material and Methods). We adopted minimum percentages of 30% identity and 40% similarity expected for IFT family members, given that these are routinely used parameters for assigning significant relationship among sequences. IFT has anterograde and retrograde components mediated by the plus (+) and minus (-) end-directed microtubule motors, kinesin-2 and cytoplasmic dynein, respectively (heavy chain and light chain) (Kozminski et al., 1995). Recently, we distinguished a few Leishmania proteins that could be related to flagellar and intraflagellar pathways, particularly the L. major putative Unc104-like kinesin (LmjF34.4260), a kinesin-2 subunit, and profilin and katanin homologues (Vasconcelos et al., 2007). Here, we present the identification of gene products that are specifically IFT-related sequences in Leishmania spp (as listed in Table 2) and that were analyzed in terms of their possible participation in IFT, a conserved process, which according to Sloboda (2005) may also provide the basic elements of a signal transduction mechanism that functions to provide the nucleus with information about the outside environment and even about the state of the flagellum itself. Thus, IFT may function as the central component of a signal transduction system that controls flagellar gene transcription (Sloboda, 2005). As originally described in Chlamydomonas, the IFT complex is composed of at least 17 different polypeptides (Cole et al., 1998), although only the homologs of the classical components (IFT88, -57, 52-, and -20) have been identified in all proposed models and also in human cells (Cole, 2003). Leishmania spp homologs to members of the IFT complex proteins have not been reported so far and one of the goals of our study was to determine whether or not they were conserved in Leishmania. Using a combination of bioinformatics tools, we report here the consistent presence of these homologs in two Leishmania genomes (Table 2), together with their sequence comparisons and structural/functional predictions. Our attempt to unveil the IFT pathway in trypanosomatids, through the annotation and analyses of gene products in their genomes, makes this survey a contribution to the formal identification of Leishmania homologues to these IFT proteins.
Subunits of the intraflagellar transport complex A in Leishmania spp - IFT140 In C. reinhardtii, the IFT complex A is said to be a 550-kDa tetramer containing six subunits, although only four of these have been clearly, unambiguously, isolated by all workers in the field (IFT144, IFT140, IFT139, and IFT122); the other two complex A subunits (IFT43 and IFT148) have not yet been confirmed unanimously by the different groups (Piperno and Mead, 1997; Piperno et al., 1998; Rosenbaum et al., 1999; Deane et al., 2001; Cole, 2003; Pazour et al., 2005; Efimenko et al., 2006; Blacque et al., 2006). A weak self-association of complex A and a weak association between complex A and B are known to occur (Cole et al., 1998). In our survey, the only component of the complex A that we were able to detect was IFT140; two sequences (GeneDB IDs: LmjF32.0310 and LinJ32.0680) displayed a significantly high similarity to the C. reinhardtii IFT140 subunit (accession No. AAT95430): 38 and 41% identities, as well as 57 and 61% similarities, for LinJ32.0680 and LmjF32.0310, respectively (Figure 1). The complete sequence of IFT140 genes in L. major and L. infantum (annotated as conserved, hypothetical proteins) encodes a 1655-residue protein with a calculated molecular mass of 181.2 kDa and an Ip of 5.9 (Table 2). Full-length IFT140 in Chlamydomonas is 1384 amino acids long, and the protein is known to have a typical WD-40 repeat domain presenting five copies of the repeat. Leishmania IFT140 (Figure 1) possesses three copies of WD-40. The WD-40 domain is found in a number of eukaryotic proteins that cover a wide variety of functions, including adaptor/regulatory modules in signal transduction, pre-mRNA processing and cytoskeleton assembly. It contains a GH dipeptide 11-24 residues from its N-terminus and the WD dipeptide at its C-terminus, and it is 40 residues long, hence the name WD-40 (Smith et al., 1999; Marchler-Bauer et al., 2005). Subunits of the intraflagellar transport complex B in Leishmania spp IFT complex B is a 750-kDa complex containing subunits ranging from 20 to 172 kDa. There are 11 (IFT20, IFT27, IFT46, IFT52, IFT57, IFT74/72, IFT80, IFT81, IFT88, and IFT172) well-characterized subunits of complex B (Cole, 2003), some of which can be dissociated to reveal a more stable set of proteins termed the subcomplex B core, distinct from a subcomplex B periphery (Lucker et al., 2005). Since IFT74 and IFT72 are nearly identical and are encoded by the same gene, they are often referred to as IFT74/72 (Qin et al., 2004). Complex B subunits usually display prominent WD-40 and TPR (tetratricopeptide repeats), among other conserved domains. WD-40-containing proteins are thought to fold into a β-propeller structure and to coordinate multiprotein complex assemblies (Smith et al., 1999). IFT particle assembly could serve as an example for such complexes. TPR motifs occur as 3-16 tandem repeats per protein that are packed in a parallel manner and form a superhelical structure for interaction with a diverse range of target proteins (D’Andrea and Regan, 2003).
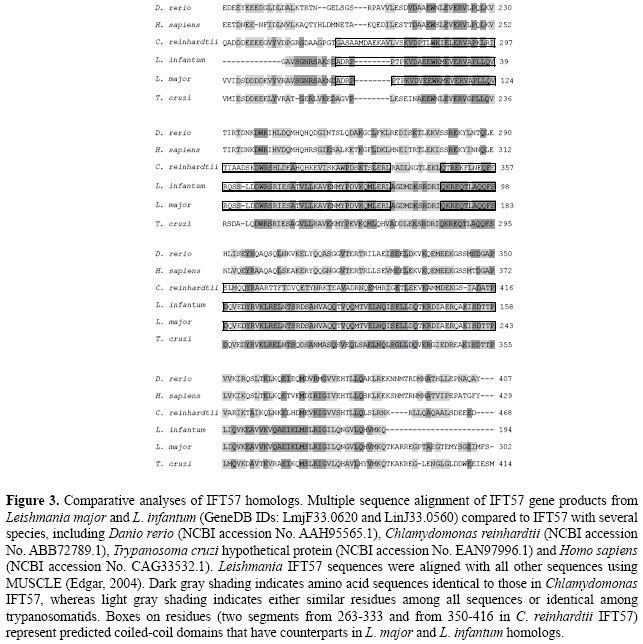
Leishmania IFT20 and IFT57 homologues In both L. major and L. infantum genomes, there are sequences that we could identify as putative IFT20 genes: one hypothetical protein (GeneDB ID: LinJ30.1860) and two unnumbered, IFT proteins (GneDB IDs: LmjF30.2000 and LinJ30.2000). The complete sequences of these latter genes encode a predicted 130-residue protein with a calculated molecular mass of 15.4 kDa and an Ip of 4.6. They have an overall range of 30-39% identity to the respective homolog in C. reinhardtii (UniProt ID: Q8LLV9) and 70-82% identity with T. brucei and T. cruzi orthologs (Figure 2). The hypothetical protein LinJ30.1860 is, however, a different case, since it has a predicted 485-residue length with a calculated molecular mass of 51.5 kDa and an Ip of 7.4. Despite this, the primary sequence of LinJ30.1860 aligns with an overlapping region of LmjF30.2000 and LinJ30.2000 (data not shown). Considering that the overall difference in extension (LinJ30.1860 is more than three times longer than a canonical IFT20 sequence) and considering such divergent predicted molecular features, we chose not to assign a regular IFT subunit name to it at this time. However, this assignment is more reasonable for the other two gene products (GeneDB IDs: LmjF30.2000 and LinJ30.2000). Figure 2 shows the multiple sequence alignments of the predicted Leishmania IFT20 proteins, compared to several characterized IFT20 proteins (from murine, bovine, human, C. reinhardtii, C. elegans) and to the ortholog in T. brucei (GeneDB ID: Tb06.2N9.700). Both Leishmania putative IFT20 proteins of 130 amino acid residues share high similarity when compared to IFT20 from diverse organisms, including mammalian homologs. A segment encompassing residues 43-117 is predicted as a coiled-coil domain believed to be an interaction site with IFT57, as reported by Baker et al. (2003). The Unc104-kinesin homolog (GeneDB ID: LmjF34.4260) in Leishmania genome illustrates a subunit of heterotrimeric kinesin-2, possibly as part of the IFT complex in an ATP-regulated manner. IFT20 appears to function in bridging the two complexes by directly interacting with both IFT57 and KIF3B/Unc104 (Baker et al., 2003). In our searches, we could find a sequence in both Leishmania genomes (GeneDB IDs: LmjF33.0620 and LinJ33.0560). The sequences showed high similarity to the IFT57 homolog in C. reinhardtii, as depicted by alignments in Figure 3. Leishmania major IFT57 identified in our study (Table 1) has a predicted protein length of 302 amino acids with a calculated molecular mass of 33.8 kDa and Ip of 4.4. The full-length IFT57 in Chlamydomonas is 429 amino acids, and in Danio rerio it is 407 residues, with a calculated molecular mass of 46.5 kDa. A secondary structure analysis of IFT57 was able to predict two immediately adjacent coiled-coil domains near the C terminus, previously identified as a myosin-like domain and a pseudo-death effector domain (Baker et al., 2003). We were able to predict the same about the Leishmania IFT57 homologs, due to their high amino acid identity in these adjacent coiled-coil domains (Figure 3). The entire extended coiled-coil region, including the pseudo-death effector domain in IFT57, seems to be required for optimal interaction with IFT20, as reported by Baker et al. (2003), another line of reasoning to be considered in our functional assignments.
Leishmania IFT52 and IFT88 homologues We identified one copy of putative IFT52 genes in both Leishmania genomes (GeneDB IDs: LmjF19.0320 and LinJ19.0120) and IFT88 (GeneDB IDs: LmjF27.1130 and LinJ27.1180) (Table 2). The complete sequence of IFT52 genes in L. major and L. infantum encodes a 672-residue protein with a calculated molecular mass of 73.2 kDa and an Ip of 4.9. Leishmania IFT52 gene products have 40-46% identity and 62-57% similarity to the respective homolog in C. reinhardtii (UniProt ID: Q944U2) along the 672 amino acid length of the predicted proteins (Figure 4). Their significant high identities (over 80%) with T. brucei and T. cruzi orthologs (GeneDB IDs: Tb10.61.1590 and Tc00.1047053506211.40) help to reinforce their identification in this study. Chlamydomonas IFT52 is 49% identical to a rodent protein called NGD5 and to a C. elegans protein called OSM-6 (Cole et al., 1998; Deane et al., 2001), whereas L. major IFT52 is 35.9% identical to murine NGD5 and 36.9% identical to C. reinhardtii OSM-6 (UniProt ID: Q946G4), the latter a gene required for flagellar assembly in Chlamydomonas (Brazelton et al., 2001) and for assembly of sensory cilia in nematodes (Cole et al., 1998). Many mammalian and worm homologs of IFT subunits have been identified recently, while several lines of evidence suggest important functional roles for IFT in ciliated/flagellated mammalian cells (Baker et al., 2003; Cole, 2003).
The complete sequence of the putative IFT88 genes in L. major and L. infantum (GeneDB IDs: LmjF27.1130 and LinJ27.1180) encodes an 811-residue protein with a calculated molecular mass of 91.2 kDa and an Ip of 6.5. The Leishmania IFT88 proteins were aligned with homologs in C. reinhardtii, T. brucei and D. rerio for comparison (Figure 5A). The analysis revealed typical TPR domains, with a variable number of nine for LmjF27.1130 and 10 for LinJ27.1180, as opposed to the normal 10 TPR motifs in C. reinhardtii (UniProt ID: Q9FPWO) and 11 in T. brucei (UniProt ID: AAP80732) (Figure 5B and C). There was a similar pattern of protein architecture exhibited by Leishmania putative IFT88 homologs, when compared to the well-established IFT88 proteins (in Chlamydomonas and D. rerio). The amino acid identity and the conservation of functional domains of IFT88 homologs extend throughout the entire length of the Leishmania 811-residue protein, which is slightly longer than the 782-residue protein in C. reinhardtii IFT88. The similarity between the Leishmania and the green alga protein throughout the full extent of the latter is also apparent in four regions of particularly high similarity, all with at least 78% amino acid identity, encompassing a segment (residues 327-375 in C. reinhardtii or 332-377 in L. infantum) that includes nearly all residues of a site known to interact with proline-rich segment-containing proteins, such as BAT2, as predicted by conserved domain databases (Marchler-Bauer et al., 2005).
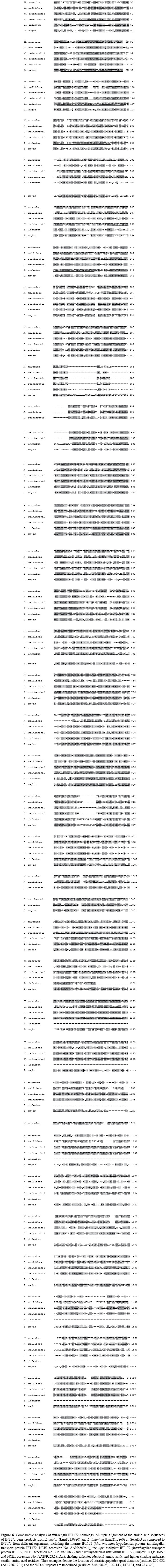
Putative IFT172 homologs IFT172 has been characterized as a typical WD-40 repeat protein, in which TPR motifs are also present. The C. reinhardtii IFT172 (UniProt ID: Q5DM57) is one of the longest proteins of the IFT complex (with a 1755-residue length and a calculated molecular mass of 197.6 kDa). The L. major IFT172 homolog that we found in our searches (annotated as conserved hypothetical protein, GeneDB ID: LmjF21.0980) has a predicted length of 1801 amino acids, with a calculated molecular mass of 200.0 kDa and an Ip of 6.4. The L. major IFT172 sequence has 44% identity and 57% similarity to the respective homolog in C. reinhardtii, as can be seen in the full alignment of IFT172 homologs in Figure 6. It displays the highest similarity exactly along the residues that are believed to confer activity to the functional domains of the characterized protein in C. reinhardtii (the TPR domains and the WD-40 repeats), as seen in Figure 7. The L. infantum IFT172 homologue (another conserved hypothetical protein, GeneDB ID: LinJ21.0860) has a predicted protein length of 1163 amino acids, and it also has an overall 44% identity with a higher (66%) similarity to the respective homolog in C. reinhardtii (UniProt ID: Q5DM57).
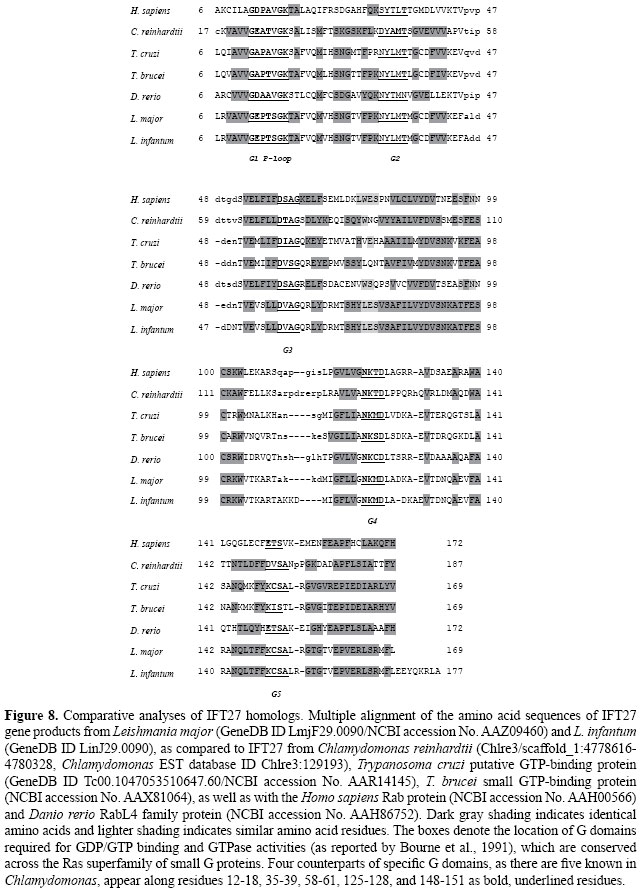
Leishmania IFT27 and IFT46 homologues IFT27 has been characterized as a Rab-like small G protein (Lucker et al., 2005) believed to be instrumental in maintaining the stability of both IFT complexes. In addition to its role in flagellar assembly, it appears to be unique among IFT polypeptides, in that its partial knockdown results in defects in cytokinesis and elongation of the cell cycle; a more complete knockdown is lethal, as recently reported by Qin et al. (2007). Based on their study, along with other studies about Neks (NIMA-related expressed kinases) and polycystins (Bradley and Quarmby, 2005), IFT27 is among the first ciliary/flagellar proteins known to be involved in cell-cycle control. The IFT27 sequence was annotated as the FAP156 gene in the C. reinhardtii genome and the flagellar proteome (Pazour et al., 2005). We used the downloaded sequence from Chlre3/scaffold_1:4778616-4780328 to perform a search on Leishmania genomes at GeneDB and found two putative homologs annotated as GTP-binding protein-like proteins: LmjF29.0090 and LinJ29.0090, with 37-38 and 52-53% identity and similarity, respectively. The full alignment of Leishmania IFT27 homologues with T. cruzi, T. brucei, D. rerio, H. sapiens, and C. reinhardtii can be seen in Figure 8, which shows that Leishmania IFT27 homologs contain at least four of the five Ras-GTPase consensus sequences that are essential for GDP/GTP binding and GTPase activity (Bourne et al., 1991), despite their relatively shorter sequences (186 amino acids, as opposed to 214 amino acids in C. reinhardtii). Generally, Rab proteins associate with cellular membranes through a prenyl (geranylgeranyl) group that is added after translation to a C-terminal prenylation motif containing one or (more frequently) two cysteine residues. Chlamydomonas IFT27 contains a prenylation motif (CRNY); however, none of the IFT27 orthologs in zebrafish, mouse, or human contain such a motif (Qin et al., 2007), and Leishmania and T. cruzi sequences lack this C-terminal motif, as demonstrated by their shortness, with about 28 residues. Concerning IFT46, it has recently been reported, through analysis of two-hybrid yeast assays, that IFT46 and IFT52 are able to interact (Lucker et al., 2005), suggesting a more direct role with IFT88 in its cargo function. We identified at least one copy of a putative IFT46 gene in Leishmania genomes (GeneDB IDs: LmjF30.1770 and LinJ302110), which shows 38.9% identical and 55% similar residues along the 316 amino acid length of the Chlamydomonas IFT46 sequence (NCBI accession No. ABHO6907.1). The alignments are shown in Figure 9, which illustrates the absence of any conserved domains or motifs; however, the overall similarity among compared sequences is enough to assign an IFT subunit number to Leishmania gene products LmjF30.1770 and LinJ302110. Other intraflagellar transport subunits (IFT71, IFT74/72 and IFT81 homologues) When compared to the subunits analyzed above, which showed more consistent similarities and amino acid identities, there were a few other sequences that we could also distinguish as putative IFT subunits. Such sequences, showing an overall lower similarity (but still significant) to other components of the IFT complex, could also be found in Leishmania genomes. Among them, there are two intraflagellar transport protein-like (GeneDB IDs: LmjF22.1370 protein/CHR22_tmp.1270 and LinJ35.1580) genes that showed similarity; they showed 22% identity and 51% similarity to IFT71 subunit (UniProt ID: Q6RCE1) and 23% identity and 51% similarity to the IFT74/72 subunit (UniProt ID: Q84P51) of C. reinhardtii. In addition, we observed that the hypothetical proteins (GeneDB IDs: LmjF34.0230 and LinJ34.0220) showed 25% identity and 49% similarity to the C. reinhardtii IFT81 subunit (UniProt ID: Q68RJ5) and 30/58% to the D. rerio IFT81 (accession No. AAT39118). These observations are consistent with reports that clarify the existence of the IFT complex B core or subcomplex, consisting of a more stable set of proteins that includes IFT81, IFT74, and IFT72 (Lucker et al., 2005). Combining two-hybrid and three-hybrid analyses, Lucker et al. (2005) showed that IFT81 can be routinely cross-linked to either IFT74 or IFT72, whereas they were unable to distinguish between these two nearly identical proteins. They demonstrated how IFT81 and IFT74/72 interact directly to form a higher order oligomer. We also made detailed alignments and respective analyses of Leishmania genes homologous to these proteins (IFT81 and IFT74/72) (data not shown); there was enough similarity to assign them as complex B subunits (Table 2).
Biological significance of the intraflagellar transport complex in Leishmania spp A question of attributing functionality to sequence-conserved homologues The presence of so many IFT subunits in the few Leishmania genomes screened until now indicates that, although an IFT complex still needs to be confirmed in Leishmania, it is likely to be similar to the one seen in Chlamydomonas. With the advances in genome-related research and the computational biology advent, whose analyses have fundamentally changed the nature of research strategies, there has been an explosion of new information on all types of proteins, including the actin-related and flagellar-associated proteins, their regulation, their roles in signaling and also in flagellar assembly and disassembly. Some of these proteins have close homologs in prokaryotic and eukaryotic systems, indicating that the mechanisms behind these flagellar mechanisms are probably similar across divergent species (Cole, 2003). The observation that most IFT complex components have counterparts in Leishmania genomes (Table 2) provides insight into the possible interactions between the IFT complex subunits and actin-related proteins, such as kinesin-2, reported to be involved with the assembly and maintenance of all cilia and flagella in eukaryotic cells (Cole et al., 1998). Knowledge about these flagellar proteins in Leishmania might support the recent suggestion that kinesin-2 is needed in cell fusion in order to localize and transport flagellar agglutinins (Rosenbaum et al., 1999; Iomini et al., 2001). Our understanding of their significance remains limited, because they have not been genetically characterized regarding trypanosomatid flagellar activity. The function of complexes A and B, as well as the contribution of basal bodies and distal structures of flagella (Dentler and Rosenbaum, 1977) to IFT mechanisms, have not been identified, not even in the advanced studies on Chlamydomonas. Although significant strides have been made in dissecting the mechanisms of IFT, it remains a poorly understood process. For instance, the full complement of its components is unknown, and the organization, regulation, and specific functions of the IFT machinery are incompletely understood (reviewed by Blacque et al., 2006). It is widely accepted that gene function can be predicted by identifying, in silico, pairs of genes whose evolution is correlated between organisms, or whose homologs are fused into a single gene in other organisms (Koonin and Galperin, 2002). Although many genomes have been sequenced, the precise identification of genes that are expressed is still a work in progress for most organisms, because the sequence features that govern transcription, splicing and translation are not fully understood (reviewed by Carpenter and Sabatini, 2004). However, functional categorization of genes that are found in genome screenings (as we have done here) can demonstrate regulators of (or contributors to) cellular processes, as we have attempted for IFT in Leishmania. Using a simple combination of computer methods for interactive database searches and refined multiple sequence alignments, we showed that gene products related to IFT in Leishmania spp are highly conserved and can be regarded as components of the IFT complex. Considering the very high sequence homology and structural similarities among the putative Leishmania and Trypanosoma orthologs of IFT subunits and a number of other IFT complex subunits from several species (Figures 2-5,7,9), we suggest that they are functional homologs of IFT-complex components. ACKNOWLEDGMENTS Research supported by Brazilian research funding agencies CNPq, FINEP, BNB/FUNDECI, and FUNCAP through individual grants to D.M. Oliveira. A.C.L. Pacheco, E.J.R. Vasconcelos, J.J.S. Gouveia, M.P. Costa, D.A. Viana, and A.R.S. Maia were recipients of FUNCAP and CNPq fellowhips. REFERENCES Acestor N, Masina S, Walker J, Saravia NG, et al. (2002). Establishing two-dimensional gels for the analysis of Leishmania proteomes. Proteomics 2: 877-879. Aggarwal G, Worthey EA, McDonagh PD and Myler PJ (2003). Importing statistical measures into Artemis enhances gene identification in the Leishmania genome project. BMC Bioinformatics 4: 23. Alfarano C, Andrade CE, Anthony K, Bahroos N, et al. (2005). The biomolecular interaction network database and related tools 2005 update. Nucleic Acids Res. 33: D418-D424. Altschul SF, Madden TL, Schaffer AA, Zhang J, et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, et al. (2004). Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117: 527-539. Baker SA, Freeman K, Luby-Phelps K, Pazour GJ, et al. (2003). IFT20 links kinesin II with a mammalian intraflagellar transport complex that is conserved in motile flagella and sensory cilia. J. Biol. Chem. 278: 34211-34218. Blacque OE, Li C, Inglis PN, Esmail MA, et al. (2006). The WD repeat-containing protein IFTA-1 is required for retrograde intraflagellar transport. Mol. Biol. Cell 17: 5053-5062. Bourne HR, Sanders DA and McCormick F (1991). The GTPase superfamily: conserved structure and molecular mechanism. Nature 349: 117-127. Bradley BA and Quarmby LM (2005). A NIMA-related kinase, Cnk2p, regulates both flagellar length and cell size in Chlamydomonas. J. Cell Sci. 118: 3317-3326. Brazelton WJ, Amundsen CD, Silflow CD and Lefebvre PA (2001). The bld1 mutation identifies the Chlamydomonas osm-6 homolog as a gene required for flagellar assembly. Curr. Biol. 11: 1591-1594. Carpenter AE and Sabatini DM (2004). Systematic genome-wide screens of gene function. Nat. Rev. Genet. 5: 11-22. Cole DG (2003). The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic 4: 435-442. Cole DG, Diener DR, Himelblau AL, Beech PL, et al. (1998). Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J. Cell Biol. 141: 993-1008. D’Andrea LD and Regan L (2003). TPR proteins: the versatile helix. Trends Biochem. Sci. 28: 655-662. Deane JA, Cole DG, Seeley ES, Diener DR, et al. (2001). Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr. Biol. 11: 1586-1590. Dentler WL and Rosenbaum JL (1977). Flagellar elongation and shortening in Chlamydomonas. III. Structures attached to the tips of flagellar microtubules and their relationship to the directionality of flagellar microtubule assembly. J. Cell Biol. 74: 747-759. Drummelsmith J, Brochu V, Girard I, Messier N, et al. (2003). Proteome mapping of the protozoan parasite Leishmania and application to the study of drug targets and resistance mechanisms. Mol. Cell Proteomics 2: 146-155. Drummelsmith J, Girard I, Trudel N and Ouellette M (2004). Differential protein expression analysis of Leishmania major reveals novel roles for methionine adenosyltransferase and S-adenosylmethionine in methotrexate resistance. J. Biol. Chem. 279: 33273-33280. Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32: 1792-1797. Efimenko E, Blacque OE, Ou G, Haycraft CJ, et al. (2006). Caenorhabditis elegans DYF-2, an orthologue of human WDR19, is a component of the intraflagellar transport machinery in sensory cilia. Mol. Biol. Cell 17: 4801-4811. El Fakhry Y, Ouellette M and Papadopoulou B (2002). A proteomic approach to identify developmentally regulated proteins in Leishmania infantum. Proteomics 2: 1007-1017. Emanuelsson O, Nielsen H, Brunak S and von Heijne G (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300: 1005-1016. Ersfeld K and Gull K (2000). Targeting of cytoskeletal proteins to the flagellum of Trypanosoma brucei. J. Cell Sci. 114: 141-148. Haycraft CJ, Schafer JC, Zhang Q, Taulman PD, et al. (2003). Identification of CHE-13, a novel intraflagellar transport protein required for cilia formation. Exp. Cell Res. 284: 251-263. Iomini C, Babaev-Khaimov V, Sassaroli M and Piperno G (2001). Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. J. Cell Biol. 153: 13-24. Johnson KA and Rosenbaum JL (1992). Polarity of flagellar assembly in Chlamydomonas. J. Cell Biol. 119: 1605-1611. Koonin EV and Galperin MY (2002). Sequence-Evolution-Function. Computational Approaches in Comparative Genomics. Kluwer Academic Publishers, Boston. Kozminski KG, Beech PL and Rosenbaum JL (1995). The Chlamydomonas kinesin-like protein Fla10 is involved in motility associated with the flagellar membrane. J. Cell Biol. 131: 1517-1527. Letunic I, Copley RR, Pils B, Pinkert S, et al. (2006). SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 34: D257-D260. Lucker BF, Behal RH, Qin H, Siron LC, et al. (2005). Characterization of the intraflagellar transport complex B core: direct interaction of the IFT81 and IFT74/72 subunits. J. Biol. Chem. 280: 27688-27696. Majoros WH, Pertea M and Salzberg SL (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20: 2878-2879. Marchler-Bauer A, Anderson JB, Cherukuri PF, Weese-Scott C, et al. (2005). CDD: a conserved domain database for protein classification. Nucleic Acids Res. 33: D192-D196. Oliveira DM, Gouveia JJ, Diniz NB, Pacheco AC, et al. (2005). Pathogenomics analysis of Leishmania spp.: flagellar gene families of putative virulence factors. OMICS 9: 173-193. Parker JD and Quarmby LM (2003). Chlamydomonas fla mutants reveal a link between deflagellation and intraflagellar transport. BMC Cell Biol. 4: 11. Pazour GJ, Agrin N, Leszyk J and Witman GB (2005). Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 170: 103-113. Piperno G and Mead K (1997). Transport of a novel complex in the cytoplasmic matrix of Chlamydomonas flagella. Proc. Natl. Acad. Sci. USA 94: 4457-4462. Piperno G, Mead K and Henderson S (1996). Inner dynein arms but not outer dynein arms require the activity of kinesin homologue protein KHP1(FLA10) to reach the distal part of flagella in Chlamydomonas. J. Cell Biol. 133: 371-379. Piperno G, Siuda E, Henderson S, Segil M, et al. (1998). Distinct mutants of retrograde intraflagellar transport (IFT) share similar morphological and molecular defects. J. Cell Biol. 143: 1591-1601. Qin H, Diener DR, Geimer S, Cole DG, et al. (2004). Intraflagellar transport (IFT) cargo: IFT transports flagellar precursors to the tip and turnover products to the cell body. J. Cell Biol. 164: 255-266. Qin H, Wang Z, Diener D and Rosenbaum J (2007). Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr. Biol. 17: 193-202. Rosenbaum JL and Witman GB (2002). Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 3: 813-825. Rosenbaum JL, Cole DG and Diener DR (1999). Intraflagellar transport: the eyes have it. J. Cell Biol. 144: 385-388. Sherwin T and Gull K (1989). The cell division cycle of Trypanosoma brucei brucei: timing of event markers and cytoskeletal modulations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 323: 573-588. Sloboda RD (2005). Intraflagellar transport and the flagellar tip complex. J. Cell Biochem. 94: 266-272. Smith TF, Gaitatzes C, Saxena K and Neer EJ (1999). The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 24: 181-185. Tull D, Vince JE, Callaghan JM, Naderer T, et al. (2004). SMP-1, a member of a new family of small myristoylated proteins in kinetoplastid parasites, is targeted to the flagellum membrane in Leishmania. Mol. Biol. Cell 15: 4775-4786. Vasconcelos EJR, Pacheco ACL, Gouveia JJS, Araújo FF, et al. (2007). Profilins, formins and katanins as flagellar proteins of Leishmania ssp.: a genome-based, multi-step bioinformatics-driven description. In: IEEE 7th International Symposium on Bioinformatics and Bioengineering. Edited by Bourbakis NG et al., IEEE BIBE 2007 Conference Proceedings, Boston. von Mering C, Jensen LJ, Snel B, Hooper SD, et al. (2005). STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33: D433-D437. |
|