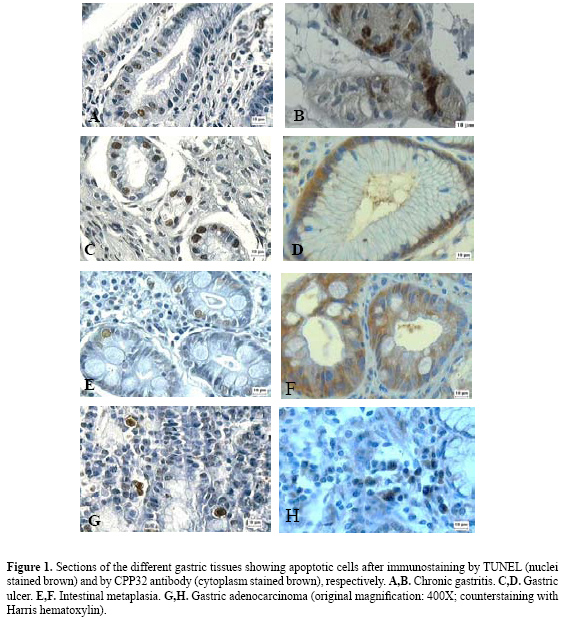
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
| Apoptosis in different gastric lesions and gastric cancer: relationship with Helicobacter pylori, overexpression of p53 and aneuploidy A.C. Targa1, A.C.G. César2, P.M. Cury3 and A.E. Silva1 1Departamento de Biologia, Universidade Estadual Paulista, UNESP, São José do Rio Preto, SP, Brasil 2Universidade Paulista, UNIP, Araçatuba, SP, Brasil 3Departamento de Patologia, Faculdade de Medicina, FAMERP, São José do Rio Preto, SP, Brasil Corresponding author: A.E. Silva E-mail: [email protected] Genet. Mol. Res. 6 (3): 554-565 (2007) Received May 5, 2007 Accepted August 24, 2007 Published September 30, 2007 ABSTRACT. Apoptosis has an essential function in maintaining the integrity of the gastrointestinal mucosa. Its deregulation is associated with the occurrence of lesions such as in atrophic gastritis, peptic ulcers, intestinal metaplasia, and stomach tumorigenesis. Thus, the aim of the present study was to investigate the frequency of apoptotic cells (apoptotic index, AI) by using two different immunohistochemical techniques, TUNEL and anti-activated caspase-3 antibody (CPP32), in gastric dyspepsia [chronic gastritis (CG, N = 34), chronic atrophic gastritis (CAG, N = 11), gastric ulcer (GU, N = 17), and intestinal metaplasia (IM, N = 15)], normal gastric mucosae (NM, N = 8), and gastric adenocarcinoma (GC, N = 12). The relationship was investigated between the AI and Helicobacter pylori infection, diagnosed by PCR, overexpression of p53 protein determined by immunohistochemistry, and aneuploidy by fluorescence in situ hybridization, as performed by our laboratory in previous studies. No significant differences were observed in AI between the different groups, whether by the TUNEL technique (F = 1.60; p = 0.1670) or by CPP32 antibody (F = 1.70; p = 0.1420). Nonetheless, CAG and CG groups had AI statistically higher than those of normal mucosae. These two groups (CAG and CG) also showed a higher frequency of apoptosis-positive cases (TUNEL+ or CPP32+). Generally, there was no correlation between the AI detected by the TUNEL and CPP32 techniques in the groups studied, except in the GC group (r = 0.70). Moreover, there was no significant association between apoptosis and H. pylori infection, overexpression of p53 protein and aneuploidy, but the H. pylori-positive cases only of GU (p = 0.0233) and IM (p = 0.0253) groups displayed a statistically higher AI compared to H. pylori-negative NM, when the CPP32 antibody technique was used. Thus, CG and CAG have increased apoptosis, which may occur independent of an association with H. pylori infection, aneuploidy and overexpression of p53 protein. Key words: Apoptosis, Helicobacter pylori, Activated caspase-3, INTRODUCTION Gastric epithelial homeostasis is maintained by a balance between cell proliferation rate and programmed cell death or apoptosis. An imbalance of these two processes leading to increased proliferation of the gastric mucosa may enhance the effect of carcinogens on DNA, increasing the risk of mutational changes and the development of gastric cancer (Yang et al., 2003; Naumann and Crabtree, 2004). On the other hand, apoptosis, a genetically regulated mechanism of cell death, plays an essential role in eliminating unnecessary or damaged cells from the organism (Dlamini et al., 2004). In the multistep process of gastric carcinogenesis (GC), the bacterium Helicobacter pylori plays a major etiological role and has recently been classified as a type-I carcinogen (IARC, 1994). H. pylori infection is generally believed to be the initial trigger of the cascade of this process, beginning with a chronic active inflammatory response of the mucosae and subsequently progressing to gastric atrophy, intestinal metaplasia (IM), dysplasia and ultimately to cancer (Correa, 2004). Several studies have shown a relationship between H. pylori infection and an increased apoptosis rate in gastric lesions such as chronic gastritis (CG), generally accompanied by glandular atrophy, gastric ulcer (GU) and IM (Attallah et al., 1996; Moss et al., 1996; Steininger et al., 1998; Hirasawa et al., 1999; Nardone et al., 1999; von Herbay and Rudi, 2000; Lee et al., 2003; van Grieken et al., 2003; Ashktorab et al., 2004; Wambura et al., 2004). However, the results are divergent, because there are also studies showing that the presence or absence of infection does not influence either the apoptotic index (AI; Anti et al., 1998; de Freitas et al., 2004) or the degree of atrophy of the gastric mucosa (Anti et al., 1998). Moreover, the H. pylori infection may cause a decrease of apoptotic activity, followed by an increase of cell proliferation (Wagner et al., 1997). Eradication of H. pylori infection results in a significant decrease in gastric epithelial cell proliferation and apoptosis (Satoh et al., 2003), indicating that the increase of apoptosis in active H. pylori infection is a reversible phenomenon (von Herbay and Rudi, 2000). Apoptosis is an asynchronous process involving few cells at a specific site of the tissue examined, which may be found at different steps of this process (von Herbay and Rudi, 2000). Thus, it is important to employ more than one independent method, to allow the detection of the apoptotic cells at different stages in the tissue (Stadelmann and Lassmann, 2000; Schultz and Harrington Jr., 2003). The aim of this study was to determine the AI in histologically different groups of gastric tissue (normal gastric mucosae (NM), CG, chronic atrophic gastritis (CAG), GU, IM, and GC), using two different immunohistochemical techniques, the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) and anti-activated caspase-3 antibody (CPP32), which detect apoptotic cells in distinct steps of the process. The results were compared with H. pylori infection, overexpression of protein p53, and aneuploidy, as determined by our laboratory in previous studies (César et al., 2004, 2005, 2006), to help elucidate the role of apoptosis during the progression of GC. MATERIAL AND METHODS Samples A total of 97 specimens (8 H. pylori-negative NM, 34 CG, 11 CAG, 17 GU, 15 IM, and 12 GC), obtained from paraffin-embedded material, were evaluated. The mean age of the controls with nm was 37 years (range 20-66), whereas the mean ages of the patients with CG, GU, IM, and GC were 44 years (range 2-89), 60 years (range 28-83), 68 years (range 22-91), and 65 years (range 38-93), respectively. All specimens were collected at the Hospital de Base in São José do Rio Preto, SP, Brazil. The tumors were histologically classified according to Lauren (1965) as intestinal-type or diffuse-type. Biopsies of nm were obtained from individuals who had undergone endoscopic evaluation for gastric dyspepsia whose histological diagnostic confirmed normal and H. pylori-negative mucosa. The CG and IM specimens were histopathologically diagnosed and classified according to the Sydney System (Dixon et al., 1996). All nm and im specimens were collected from the antrum region of the stomach, whereas those of CG and GC were mainly collected from the antrum and corpus. The study was approved by the National Research Ethics Committee. H. pylori infection was previously diagnosed in our laboratory by PCR, utilizing the primers H3H4 and H5H6 (César et al., 2005), as was the frequency of aneuploidies of chromosomes 3, 7, 8, 9, and 17 by fluorescence in situ hybridization (FISH), and p53 expression by immunohistochemistry (César et al., 2004, 2006). Immunohistochemical analysis Apoptotic cells were detected by the TUNEL method, using a FragEL detection kit (Oncogene, Cambridge, Ma, USA) according to the manufacturer’s instructions, and by the CPP32 technique. Briefly, sections were cut at 5 µm, deparaffinized in xylene, and rehydrated with a graded ethanol series. After being washed in Tris-buffer saline (TBS, pH 7.4), the sections were placed in H2O2 for 10 min, and the tissues were then digested with proteinase K (20 µg/mL in TBS) at 37ºC for 10 min, to enhance nuclear staining of apoptotic cells. Digestion was stopped by washing the sections in TBS. The sections were then treated with terminal transferase enzyme and digoxigenin-labeled nucleotides, and afterwards an anti-digoxigenin-peroxidase solution was applied. The color was developed with DAB, after which the sections were lightly counterstained with hematoxylin. To confirm the staining specificity of the TUNEL method, a positive control section was prepared. The substitution of equilibration buffer for TdT was used as the negative control. The detection of apoptotic cells by CPP32 expression was performed using the monoclonal antibody CPP32 (Novocastra) in combination with the anti-Ig second-stage antibody and the ABC Kit (Novocastra). This antibody produces cytoplasmic staining. As positive control, intestinal mucosa sections were processed, which were previously shown to strongly express the antibody. For negative controls, the primary antibody was omitted from the buffer solution. The AI was calculated as the percentage of TUNEL-positive (nuclear immunostaining) or CPP32-positive cells (cytoplasmic immunostaining) in about 500 epithelial cells examined for each sample, under a light microscope (400X magnification). All slides were coded and scored by one observer. Areas that were poorly preserved, crushed, cauterized, folded, or retracted were specifically avoided. Statistical analysis Descriptive statistical, ANOVA, Fisher’s exact, chi-square, and Student t-tests were used to determine statistical significance. Values of P less than 0.05 were considered to be significant. The statistical analyses were performed using the Statidisk and GraphPad Instat computer software programs. RESULTS Table 1 shows the results regarding the AI by the TUNEL and CPP32 antibody methods in NM, CG, CAG, GU, IM, and GC. The sample of H. pylori-negative NM used as control did not exhibit increased apoptosis, aneuploidy or p53 overexpression, thus none of them was defined as positive. The mean frequency of immunostained apoptotic cells by the TUNEL method was 1.82 ± 0.47 (mean ± SEM; range 0-3.6%), and by the CPP32 antibody it was 3.20 ± 1.31 (range 0-11%). Thus, the cases which were considered as positive (increased ai) were those of benign lesions and GC with more than 3.6% immunostained cells by the TUNEL method and >11% immunostained cells by the CPP32 antibody technique. No significant differences were observed in the AI among the different gastric lesion groups, whether by the TUNEL technique (F = 1.60; p = 0.1670) or by the CPP32 antibody (F = 1.70; p = 0.1420). However, by the TUNEL method, the CAG group showed a statistically increased AI (3.93 ± 0.63) (p = 0.0231), compared to nm, as well as a higher number of TUNEL-positive cases (6/11, 54.5%). CPP32 antibody revealed that the CG group had a statistically higher AI (13.82 ± 2.58) than did the nm (p = 0.0311), as well as a higher number of CPP32-positive cases (15/34, 44.1%). The other groups did not show a statistically different AI compared to nm, by either of the two methods. 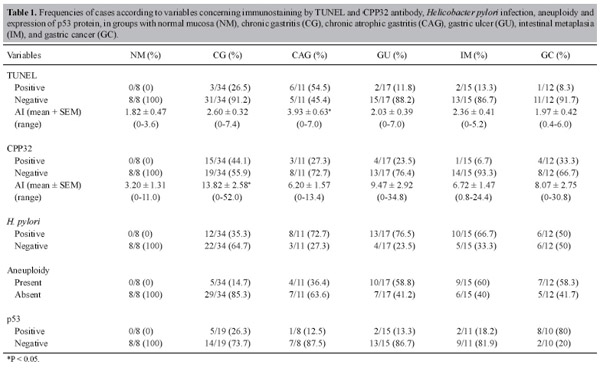
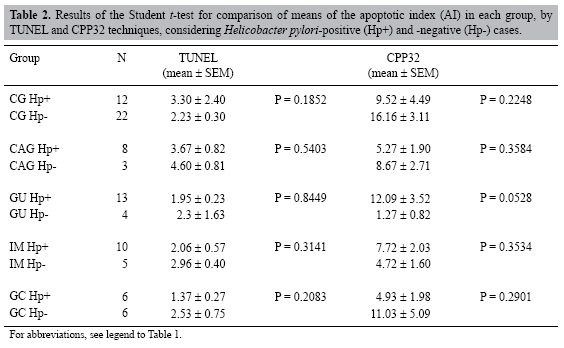
The GC group displayed a low AI, and no significant differences were found regarding the histological subtypes, intestinal or diffuse, either by the TUNEL (p = 0.2810) or by the CPP32 antibody method (p = 0.3134). Correlation analysis did not show any correlation between the AI of each case by TUNEL and antibody CPP32 in the groups of nm and benign lesions, as opposed to the GC group (r = 0.70). Figure 1 illustrates the TUNEL-positive nuclear immunostaining and the CPP32-positive cytoplasmic immunostaining, mainly in glandular and faveolar epithelium of benign lesions and in GC infiltrate. According to the Student t-test, for a comparison of the AI in each group (Table 2) with regard to H. pylori-positive or -negative cases, no significant differences were found. However, the H. pylori-positive cases of the GU (p = 0.0233) and IM (p = 0.0253) groups displayed a statistically higher AI when compared with H. pylori-negative nm, but only by the CPP32 antibody method. The Fisher exact test showed no association between the TUNEL-positive and CPP32-positive cases and either H. pylori infection, presence of aneuploidies (chromosomes 3, 7, 8, 9, and 17) or overexpression of p53 protein (data not shown). DISCUSSION Over the last decades, great advances have been made, giving insights into the molecular and cellular mechanisms involved in GC. Multiple genetic and epigenetic alterations in oncogenes, tumor suppressor genes and repair genes that control the cell cycle and apoptosis have been reported (Smith et al., 2006). Thus, changes in these groups of genes may alter the susceptibility of tumor cells to trigger apoptosis (De Luca and Iaquinto, 2004). In addition, H. pylori infection is associated with significant damage of gastric epithelial cells and increase of the apoptosis ratio. This bacterium may affect the normal balance between cell proliferation and cell death, interfering with the maintenance of the integrity of the gastric mucosa (Sugiyama and Asaka, 2004). H. pylori is able to interact with gastric epithelial cells, activating signaling pathways which are important for carcinogenesis (Oliveira et al., 2006). In the current study, the AI was determined in different groups of gastric dyspepsia and GC, using both the TUNEL and the CPP32 antibody methods. The TUNEL technique showed evidence of a statistically higher AI in CAG (3.93%) compared to NM (1.82%), while CPP32 showed an increased AI in CG (13.82%) as compared to NM (3.20%). In both the CAG and the CG groups, greater numbers of TUNEL-positive and CPP32-positive cases were also observed. Other groups of benign lesions (GU and IM) and GC did not show increased rates of apoptosis in comparison to NM. Similarly, Leite et al. (2005) evaluated different groups of patients with dyspeptic symptoms and found the highest apoptosis rate in CAG (5.2%) compared to NM (1.4%), using only the TUNEL method. In another previous study, Yoshimura et al. (2000) observed a positive correlation between the AI and the degree of glandular atrophy. Thus, the increase in apoptotic cells that is not accompanied by a matched increase in cell proliferation may play an important role in the development of glandular atrophy, consequently increasing the risk of GC.
Apoptosis of gastric epithelial cells appears to be influenced by some factors, such as variations in H. pylori pathogenicity, DNA damage, and expression of proteins that control the cell cycle and apoptosis. However, in this study, we did not observe an association between AI and either TUNEL-positive or CPP32-positive cases with H. pylori infection, aneuploidy (chromosomes 3, 7, 8, 9, and 17, detected by FISH technique) or p53 protein expression.
H. pylori infection plays a crucial role in the pathogenesis of CG, CAG, and MALT lymphoma (NIH Consensus Conference, 1994), and it is related to a 2.7- to 12-fold risk of developing GC (Cover and Blaser, 1995). Although the exact pathophysiological mechanisms of this process still need to be further elucidated, some of the bacterial virulence factors, which include VacA and CagA, host genetic susceptibility, and immune response may be involved in the process (Matysiak-Budnik and Mégraud, 2006). In addition, H. pylori infection is accompanied by an increased formation of oxygen free radicals, reactive nitrogen species and peroxidative damage, and generation of nitric oxide by inducible nitric oxide synthase, which together can increase the risk of DNA damage in proliferating cells (Yang et al., 2003; De Luca and Iaquinto, 2004; Matysiak-Budnik and Mégraud, 2006). Yet, H. pylori leads to apoptosis in the gastric epithelium through downregulation of the antiapoptotic Bcl-2, upregulation of Bax (Yang et al., 2003; Liu et al., 2005), release of cytochrome c into cytosol, and activation of caspase-9 (Potthoff et al., 2002), caspase-8, -3, and -1 (Ashktorab et al., 2002; Chen et al., 2005). Our study showed no significant difference between patients with and without H. pylori infection, in any of the groups examined. Moreover, there was no statistical difference either between the AI of the H. pylori-negative NM group and the H. pylori-positive cases of the groups with CG, CAG, and GC. Conversely, CPP32 antibody showed evidence of a higher AI in patients with GU and IM infected with H. pylori than in those with H. pylori-negative NM. While Leite et al. (2005) did not observe any statistical difference in AI between the H. pylori-positive and the H. pylori-negative groups with gastric dyspepsia, Tiwari et al. (2005) found a higher AI in the GC and non-ulcer dyspepsia groups with H. pylori infection. These contradictory results may be the consequence of virulence factors of the bacterium, such as CAG pathogenicity islands (cagPAI) and VacA (vacuolating cytotoxin), which may be responsible for the differences in apoptosis ratios reported in different groups of gastric dyspepsia and GC in patients infected with H. pylori (Naumann and Crabtree, 2004; Sugiyama and Asaka, 2004). Apoptotic studies in GU and IM are scarce and inconclusive. Our results showed a higher AI in these two types of gastric lesion with H. pylori infection compared to H. pylori-negative NM, when the CPP32 antibody was used. There are reports on duodenal ulcer (Moss et al., 1996) and GU (Satoh et al., 2003), both showing increased AI that was significantly reduced after H. pylori eradication, and demonstrating an increase of AI in GU following treatment of the infection (Hirasawa et al., 1999). However, in other studies, no differences in AI were observed, both in GU and in duodenal ulcer, whether H. pylori-positive or -negative (Kohda et al., 1999; Jang and Kim, 2000). Leung et al. (2001) demonstrated a marked difference in cell kinetics between metaplastic and non-metaplastic gastric epithelium. In IM obtained from the gastric antrum, AI was significantly lower than that of the non-metaplastic area, both infected with H. pylori. Therefore, the AI/proliferation index ratio was markedly reduced in IM when compared to the adjacent non-metaplastic area, which favors cellular accumulation and possibly neoplasm formation. Finally, after H. pylori eradication, the apoptotic activity remained unaltered in IM, while a remarkable reduction in cell proliferation was observed. Similarly, Wambura et al. (2004) compared the rates of apoptosis and proliferation in mucosa with and without IM and also demonstrated that, while antral apoptosis was significantly lower in IM, proliferation was significantly higher. However, after successful eradication, apoptosis and proliferation decreased in both the metaplastic and the non-metaplastic epithelium. Additionally, Forones et al. (2005) observed that both AI and the proliferation index increased from intestinal metaplasia to gastric cancer. Thus, cell proliferation and apoptosis are altered in H. pylori infection, leading to an imbalance in cell kinetics (Wambura et al., 2004). It is important to keep in mind that an H. pylori eradication treatment may have no preventive effects on the development of gastric cancer, once the irreversible step in the sequence from gastritis to cancer (point of no return) has been surpassed (Sugiyama and Asaka, 2004). Recent data have shown that H. pylori eradication does not completely prevent GC, evident mainly in patients with CAG and less in non-atrophic gastritis (Kuipers and Sipponen, 2006). In GC, no significant differences were observed when only the TUNEL-positive and CPP32-positive cases with H. pylori infection were considered. Moreover, no difference in AI between the intestinal type and the diffuse type was found. Similarly, some studies also showed no difference in staining pattern between intestinal- and diffuse-type gastric adenocarcinoma, employing the CPP32 antibody as an apoptosis marker (Yoo et al., 2002; van der Woude et al., 2003). However, other studies found an increased AI in patients with gastric cancer infected with H. pylori (Yoshimura et al., 2000; Forones et al., 2005; Shiotani et al., 2005; Tiwari et al., 2005), as well as a significant difference between tissues: in the antral mucosa, the AI was higher in patients with the diffuse type, while in the corpus mucosa it was higher in patients with the intestinal type (Yoshimura et al., 2000). In our study, no statistically significant association was detected between aneuploidy and overexpression of p53 and an increased AI, as evaluated by the TUNEL or the CPP32 antibody technique. Similarly, Ikeda et al. (1998) showed that there does not seem to be an association between overexpression of p53 protein and increased AI in GC. Several other studies showed an association between H. pylori infection and overexpression of p53 in gastric lesions and/or GC (Nardone et al., 1999; Ashktorab et al., 2003; Unger et al., 2003; Kodama et al., 2005), such as an association between H. pylori infection with apoptosis and increased p53 protein (Ashktorab et al., 2003; Unger et al., 2003). In addition, Attallah et al. (1996) revealed the presence of DNA-aneuploidy in CG associated with apoptosis. In this study, the TUNEL and CPP32 antibody methods were accurate in the detection of cells undergoing apoptosis. However, no correlation was observed between AI determined by two the methods, except in the GC group. A similar finding was reported previously by Hoshi et al. (1998), who also observed a correlation between AI determined by TUNEL and CPP32 antibody in GC. The absence of a correlation between the two methods found in the other groups of gastric lesions may be due to several factors. For instance, some cells may undergo apoptosis without the activation of caspase-3 (Mainwaring et al., 1998; Hadjiloucas et al., 2001). In addition, previous studies have pointed to either lack of correlation (Giannopoulou et al., 2002; Parton et al., 2002) or the presence of positive correlation (Vakkala et al., 1999; Hadjiloucas et al., 2001) between the AI detected by the two methods in different types of tumors. CPP32 antibody detects cells undergoing apoptosis at an early stage, before DNA fragmentation, a late event in the apoptotic process measured by the TUNEL assay. Despite the fact that the CPP32 method is more sensitive, specific and less dependent on the observer’s subjective assessment, we recommend the use of at least two different methods, whenever possible, which could provide complementary findings. In conclusion, our investigation showed that both chronic gastritis and atrophic gastritis have increased apoptosis which may occur independent of H. pylori infection, aneuploidy and overexpression of the p53 protein. However, the precise envolvement of H. pylori in the balance between apoptosis and proliferation yet need to be elucidated. ACKNOWLEDGMENTS We thank Sebastião Roberto Taboga, PhD, and Domingos Zanchetta Neto for their help with the immunohistochemistry technique, and José Antonio Manzato, PhD, for help with the statistical analysis. Research supported by the Brazilian Agency FAPESP (No. 02/12833-2). REFERENCES Anti M, Armuzzi A, Gasbarrini A and Gasbarrini G (1998). Importance of changes in epithelial cell turnover during Helicobacter pylori infection in gastric carcinogenesis. Gut 43 (Suppl 1): S27-S32. Ashktorab H, Neapolitano M, Bomma C, Allen C, et al. (2002). In vivo and in vitro activation of caspase-8 and -3 associated with Helicobacter pylori infection. Microbes Infect. 4: 713-722. Ashktorab H, Ahmed A, Littleton G, Wang XW, et al. (2003). p53 and p14 increase sensitivity of gastric cells to H. pylori-induced apoptosis. Dig. Dis. Sci. 48: 1284-1291. Ashktorab H, Frank S, Khaled AR, Durum SK, et al. (2004). Bax translocation and mitochondrial fragmentation induced by Helicobacter pylori. Gut 53: 805-813. Attallah AM, Abdel-Wahab M, Elshal MF, Zalata KR, et al. (1996). Apoptosis in chronic gastritis: evaluation of the gastric mucosa by DNA flow cytometry and the expression of the high molecular weight cytokeratin. Hepatogastroenterology 43: 1305-1312. Cesar ACG, Borim AA, Caetano A, Cury PM, et al. (2004). Aneuploidies, deletion, and overexpression of TP53 gene in intestinal metaplasia of patients without gastric cancer. Cancer Genet Cytogenet. 153: 127-132. César ACG, Cury PM, Payão SLM, Liberatore PR, et al. (2005). Comparison of histological and molecular diagnosis of Helicobacter pylori in benign lesions and gastric adenocarcinoma. Braz. J. Microbiol. 36: 12-16. Cesar ACG, de Freitas CM, Cury PM, Caetano A, et al. (2006). Genetic alterations in benign lesions: chronic gastritis and gastric ulcer. World J. Gastroenterol. 12: 625-629. Chen Y, Wang Y, Xu W and Zhang Z (2005). Analysis on the mechanism of Helicobacter pylori-induced apoptosis in gastric cancer cell line BGC-823. Int. J. Mol. Med. 16: 741-745. Correa P (2004). The biological model of gastric carcinogenesis. IARC Sci. Publ. 301-310. Cover TL and Blaser MJ (1995). Helicobacter pylori: a bacterial cause of gastritis, peptic ulcer disease and gastric cancer. Am. Soc. Micro. News 61: 21-26. de Freitas D, Urbano M, Goulao MH, Donato MM, et al. (2004). The effect of Helicobacter pylori infection on apoptosis and cell proliferation in gastric epithelium. Hepatogastroenterology 51: 876-882. De Luca A and Iaquinto G (2004). Helicobacter pylori and gastric diseases: a dangerous association. Cancer Lett. 213: 1-10. Dixon MF, Genta RM, Yardley JH and Correa P (1996). Classification and grading of gastritis. The updated Sydney system. International Workshop on the Histopathology of Gastritis, Houston 1994. Am. J. Surg. Pathol. 20: 1161-1181. Dlamini Z, Mbita Z and Zungu M (2004). Genealogy, expression, and molecular mechanisms in apoptosis. Pharmacol. Ther. 101: 1-15. Forones NM, Carvalho AP, Giannotti-Filho O, Lourenco LG, et al. (2005). Cell proliferation and apoptosis in gastric cancer and intestinal metaplasia. Arq Gastroenterol. 42: 30-34. Giannopoulou I, Nakopoulou L, Zervas A, Lazaris AC, et al. (2002). Immunohistochemical study of pro-apoptotic factors Bax, Fas and CPP32 in urinary bladder cancer: prognostic implications. Urol. Res. 30: 342-345. Hadjiloucas I, Gilmore AP, Bundred NJ and Streuli CH (2001). Assessment of apoptosis in human breast tissue using an antibody against the active form of caspase 3: relation to tumour histopathological characteristics. Br. J. Cancer 85: 1522-1526. Hirasawa R, Tatsuta M, Iishi H, Yano H, et al. (1999). Increase in apoptosis and decrease in ornithine decarboxylase activity of the gastric mucosa in patients with atrophic gastritis and gastric ulcer after successful eradication of Helicobacter pylori. Am. J. Gastroenterol. 94: 2398-2402. Hoshi T, Sasano H, Kato K, Yabuki N, et al. (1998). Immunohistochemistry of Caspase3/CPP32 in human stomach and its correlation with cell proliferation and apoptosis. Anticancer Res. 18: 4347-4353. IARC (1994). Infection with Helicobacter pylori. In: IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 61. Schistosomes, liver flukes and Helicobacter pylori. International Agency for Research on Cancer, Lyon, 177-241. Ikeda M, Shomori K, Endo K, Makino T, et al. (1998). Frequent occurrence of apoptosis is an early event in the oncogenesis of human gastric carcinoma. Virchows Arch. 432: 43-47. Jang TJ and Kim JR (2000). Proliferation and apoptosis in gastric antral epithelial cells of patients infected with Helicobacter pylori. J. Gastroenterol. 35: 265-271. Kodama M, Fujioka T, Murakami K, Okimoto T, et al. (2005). Eradication of Helicobacter pylori reduced the immunohistochemical detection of p53 and MDM2 in gastric mucosa. J. Gastroenterol. Hepatol. 20: 941-946. Kohda K, Tanaka K, Aiba Y, Yasuda M, et al. (1999). Role of apoptosis induced by Helicobacter pylori infection in the development of duodenal ulcer. Gut 44: 456-462. Kuipers EJ and Sipponen P (2006). Helicobacter pylori eradication for the prevention of gastric cancer. Helicobacter 11 (Suppl 1): 52-57. Lauren P (1965). The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 64: 31-49. Lee KM, Lee DS, Yang JM, Ahn BM, et al. (2003). Effect of Helicobacter pylori on gastric epithelial cell kinetics and expression of apoptosis-related proteins in gastric carcinogenesis. Korean J. Gastroenterol. 42: 12-19. Leite KR, Darini E, Canavez FC, Carvalho CM, et al. (2005). Helicobacter pylori and cagA gene detected by polymerase chain reaction in gastric biopsies: correlation with histological findings, proliferation and apoptosis. São Paulo Med. J. 123: 113-118. Leung WK, Yu J, To KF, Go MY, et al. (2001). Apoptosis and proliferation in Helicobacter pylori-associated gastric intestinal metaplasia. Aliment. Pharmacol. Ther. 15: 1467-1472. Liu HF, Liu WW, Wang GA and Teng XC (2005). Effect of Helicobacter pylori infection on Bax protein expression in patients with gastric precancerous lesions. World J. Gastroenterol. 11: 5899-5901. Mainwaring PN, Ellis PA, Detre S, Smith IE, et al. (1998). Comparison of in situ methods to assess DNA cleavage in apoptotic cells in patients with breast cancer. J. Clin. Pathol. 51: 34-37. Matysiak-Budnik T and Megraud F (2006). Helicobacter pylori infection and gastric cancer. Eur. J. Cancer 42: 708-716. Moss SF, Calam J, Agarwal B, Wang S, et al. (1996). Induction of gastric epithelial apoptosis by Helicobacter pylori. Gut 38: 498-501. Nardone G, Staibano S, Rocco A, Mezza E, et al. (1999). Effect of Helicobacter pylori infection and its eradication on cell proliferation, DNA status, and oncogene expression in patients with chronic gastritis. Gut 44: 789-799. Naumann M and Crabtree JE (2004). Helicobacter pylori-induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 12: 29-36. NIH Consensus Conference (1994). Helicobacter pylori in peptic ulcer diseases. NIH Consensus Development Panel on Helicobacter pylori in peptic ulcer disease. Jama 272: 65-69. Oliveira MJ, Costa AC, Costa AM, Henriques L, et al. (2006). Helicobacter pylori induces gastric epithelial cell invasion in a c-Met and type IV secretion system-dependent manner. J. Biol. Chem. 281: 34888-34896. Parton M, Krajewski S, Smith I, Krajewska M, et al. (2002). Coordinate expression of apoptosis-associated proteins in human breast cancer before and during chemotherapy. Clin. Cancer Res. 8: 2100-2108. Potthoff A, Ledig S, Martin J, Jandl O, et al. (2002). Significance of the caspase family in Helicobacter pylori induced gastric epithelial apoptosis. Helicobacter 7: 367-377. Satoh K, Kawata H, Tokumaru K, Kumakura Y, et al. (2003). Change in apoptosis in the gastric surface epithelium and glands after eradication of Helicobacter pylori. Dig. Liver Dis. 35: 78-84. Schultz DR and Harrington WJ Jr (2003). Apoptosis: programmed cell death at a molecular level. Semin. Arthritis Rheum. 32: 345-369. Shiotani A, Iishi H, Ishiguro S, Tatsuta M, et al. (2005). Epithelial cell turnover in relation to ongoing damage of the gastric mucosa in patients with early gastric cancer: increase of cell proliferation in paramalignant lesions. J. Gastroenterol. 40: 337-344. Smith MG, Hold GL, Tahara E and El-Omar EM (2006). Cellular and molecular aspects of gastric cancer. World J. Gastroenterol. 12: 2979-2990. Stadelmann C and Lassmann H (2000). Detection of apoptosis in tissue sections. Cell Tissue Res. 301: 19-31. Steininger H, Faller G, Dewald E, Brabletz T, et al. (1998). Apoptosis in chronic gastritis and its correlation with antigastric autoantibodies. Virchows Arch. 433: 13-18. Sugiyama T and Asaka M (2004). Helicobacter pylori infection and gastric cancer. Med. Electron Microsc. 37: 149-157. Tiwari S, Ghoshal U, Ghoshal UC and Dhingra S (2005). Helicobacter pylori-induced apoptosis in pathogenesis of gastric cancer. Indian J. Gastroenterol. 24: 193-196. Unger Z, Molnar B, Pronai L, Szaleczky E, et al. (2003). Mutant p53 expression and apoptotic activity of Helicobacter pylori positive and negative gastritis in correlation with the presence of intestinal metaplasia. Eur. J. Gastroenterol. Hepatol. 15: 389-393. Vakkala M, Lahteenmaki K, Raunio H, Paakko P, et al. (1999). Apoptosis during breast carcinoma progression. Clin. Cancer Res. 5: 319-324. van der Woude CJ, Kleibeuker JH, Tiebosch AT, Homan M, et al. (2003). Diffuse and intestinal type gastric carcinomas differ in their expression of apoptosis related proteins. J. Clin. Pathol. 56: 699-702. van Grieken NC, Meijer GA, zur Hausen A, Meuwissen SG, et al. (2003). Increased apoptosis in gastric mucosa adjacent to intestinal metaplasia. J. Clin. Pathol. 56: 358-361. von Herbay A and Rudi J (2000). Role of apoptosis in gastric epithelial turnover. Microsc. Res. Tech. 48: 303-311. Wagner S, Beil W, Westermann J, Logan RP, et al. (1997). Regulation of gastric epithelial cell growth by Helicobacter pylori: evidence for a major role of apoptosis. Gastroenterology 113: 1836-1847. Wambura C, Aoyama N, Shirasaka D, Kuroda K, et al. (2004). Cell kinetic balance in gastric mucosa with intestinal metaplasia after Helicobacter pylori eradication: 2-year follow-up study. Dig. Liver Dis. 36: 178-186. Yang Y, Deng CS, Peng JZ, Wong BC, et al. (2003). Effect of Helicobacter pylori on apoptosis and apoptosis related genes in gastric cancer cells. Mol. Pathol. 56: 19-24. Yoo NJ, Kim HS, Kim SY, Park WS, et al. (2002). Stomach cancer highly expresses both initiator and effector caspases; an immunohistochemical study. APMIS 110: 825-832. Yoshimura T, Shimoyama T, Tanaka M, Sasaki Y, et al. (2000). Gastric mucosal inflammation and epithelial cell turnover are associated with gastric cancer in patients with Helicobacter pylori infection. J. Clin. Pathol. 53: 532-536. |
|