![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
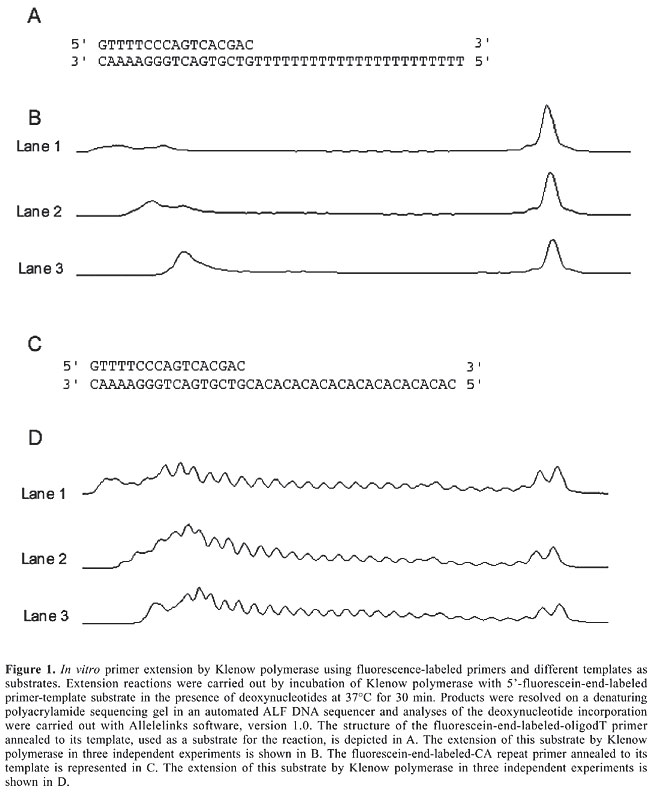
ABSTRACT. Although different DNA polymerases have distinct functions and substrate affinities, their general mechanism of action is similar. Thus, they can all be studied using the same technical principle, the primer extension assay employing radioactive tags. Even though fluorescence has been used routinely for many years for DNA sequencing, it has not been used in the in vitro primer extension assay. The use of fluorescence labels has obvious advantages over radioactivity, including safety, speed and ease of manipulation. In the present study, we demonstrated the potential of non-radioactive in vitro primer extension for DNA polymerase studies. By using an M13 tag in the substrate, we can use the same fluorescent M13 primer to study different substrate sequences. This technique allows quantification of the DNA polymerase activity of the Klenow fragment using different templates and under different conditions with similar sensitivity to the radioactive assay. Key words: Non-radioactive assay, DNA synthesis in vitro, DNA polymerase INTRODUCTION DNA-dependent DNA polymerases catalyze nucleotidyl-transfer reactions that allow all organisms to replicate their genome. On the basis of sequence homology, they are grouped into six families: A, B, C, D, X, and Y (Filee et al., 2002). Families A, B, and C were named considering the Escherichia coli genes, polA, polB, and polC, which encode the prototypical DNA polymerases I, II, and the a subunit of DNA polymerase III, respectively (Braithwaite and Ito, 1993). Family D DNA polymerases are found only in euryarchaea (1). Family X has as prototype the mammalian DNA polymerase b, a gap-filling enzyme that functions during DNA repair (Pelletier et al., 1994). Family Y encompasses the E. coli DNA polymerases IV and V, which function in the replication of DNA containing various lesions (Tippin et al., 2004). Although these different DNA polymerases present distinct functions and substrate affinities, their general mechanism of action is similar. Thus, they can all be studied using the same technical principle, the primer extension assay. The efficiency of the incorporation of nucleotide analogs (Zhang et al., 2005), DNA polymerase kinetics (Horhota et al., 2005), DNA polymerase fidelity (Roettger et al., 2004), processivity (Pavlov et al., 2006), and translesion synthesis (Yagi et al., 2005) have all been investigated with this assay, employing radioactive tags. Even though fluorescence has been for many years used routinely for DNA sequencing (Smith et al., 1987; Prober et al., 1987), it has not been used in the in vitro primer extension assay. The use of fluorescence labels has obvious advantages over radioactivity, including safety, speed and ease of manipulation. In the present study, we show the efficiency of in vitro primer extension assay using a fluorescein-end-labeled primer for DNA polymerase activity studies. MATERIAL AND METHODS To explore the use of fluorescent primers in assaying DNA polymerase activity with in vitro primer extension, we used the following 40 mer oligodeoxynucleotides as templates: an oligodT (5’ TTTTTTTTTTTTTTTTTTTTTTT GTCGTGACTGGGAAAAC) and a (dC.dA)n template (5’ CACACACACACACACACACACACGTCGTGACTGGGAAAAC), where the underlined sequences are the 17-mer M13 (-40) universal primer sequence. Fluorescein-labeled M13 primer (0.5 µM; Alpha DNA, Montreal, Canada) was annealed to the two templates (0.8 µM each) in the presence of 50 mM Tris-HCl, pH 7.5, and 100 mM NaCl by heating to 90°C for 5 min, followed by cooling to 25°C over several hours, to obtain our substrate. Since the Klenow fragment of DNA polymerase I is very well characterized, we used it as a model in our experiments. Maintaining the same conditions as in radioactive labeling primer experiments (Tuske et al., 2000; Lestienne et al., 2003), we were able to assess different settings for Klenow activity. Briefly, DNA polymerase I (Klenow fragment) was incubated with 5’-fluorescein-end-labeled primer-template substrate (50 nM), 200 µM deoxynucleotide, 10 mM Tris-HCl, pH 5, 6, 7 or 8, 5 mM dithiothreitol, 100 µg/mL bovine serum albumin, 0, 10, 100 or 200 mM MgCl2 and 0, 50, 100 or 200 mM NaCl. Reaction mixtures were incubated at 37°C for 30 min. The reaction was quenched by the addition of stop solution (10 µL) (80% deionized formamide, 10 mM EDTA, pH 8, 1 mg/mL xylene cyanol, and 1 mg/mL bromophenol blue) heated to 90°C for 2 min and placed on ice. Products were resolved on denaturing polyacrylamide sequencing gels in an automated ALF DNA sequencer (GE Healthcare). Analyses of the deoxynucleotide incorporation were carried out with Allelelinks software, version 1.0 (GE Healthcare). RESULTS AND DISCUSSION Figure 1 shows typical results with the Klenow polymerase and the two different substrates. The oligodT primer was efficiently elongated, leading to complete synthesis of all the oligodT sequence with few or no intermediate peaks (Figure 1B). However, when the CA repeat oligonucleotide was used, DNA synthesis with Klenow showed a diverse behavior: the substrate was not filled completely and intermediate peaks were evident, suggesting decrease in its processivity and therefore in its efficiency (Figure 1D).
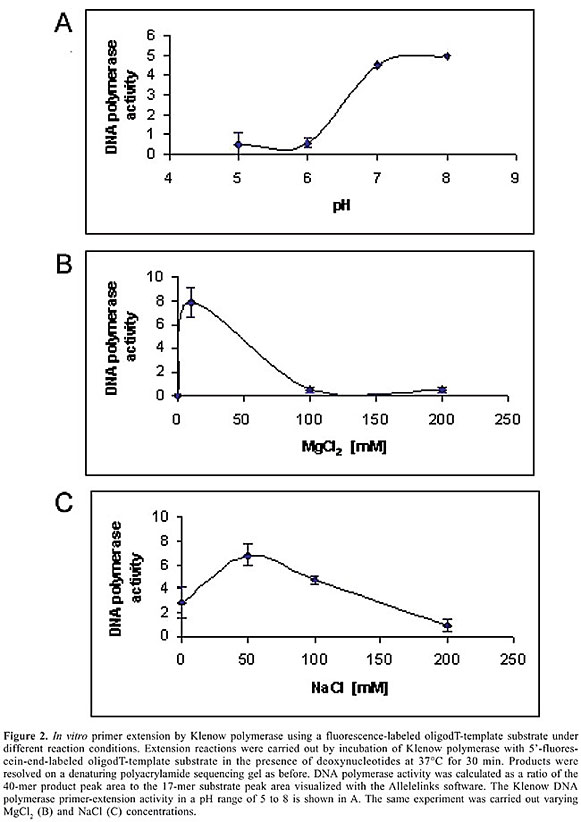
To determine if we could use the non-radioactive in vitro primer extension to quantify the DNA polymerase activity, we tested different buffer conditions using the oligodT template. The polymerase activity was calculated by dividing the 40-mer product peak area by the 17-mer substrate peak area. This procedure allows standardization of the experiments, regardless of the amount of primer used in different experiments. The data presented in this study are the average of three independent experiments. Firstly, we assessed the ideal pH for Klenow activity. Within a pH range from 5.0 to 8.0, pH 8.0 proved to be the best condition for oligodT template in our assay (Figure 2A). Indeed, this is the optimal pH recommended by the manufacturer (New England Biolabs). Secondly, we tested the MgCl2 concentration. The recommended MgCl2 concentration of 10 mM also resulted in the best activity (Figure 2B). The Klenow activity is totally abolished when no MgCl2 is added. When the MgCl2 concentration is increased 10- or 20-fold, DNA synthesis is affected and both the efficiency and activity of the Klenow polymerase decreased (Figure 2B). Finally, the best salt concentration was determined. NaCl concentration to reach Klenow’s best performance was 50 mM and higher concentrations of salt caused a decrease in enzymatic activity (Figure 2C). Thus, by using the non-radioactive in vitro primer extension, we could determine the best conditions for Klenow activity. All of them are in agreement with the manufacturer’s recommendation.
In this study we demonstrated the potential of non-radioactive in vitro primer extension for DNA polymerase studies. By using an M13 tag in the substrate, we can use the same fluorescent M13 primer to study different substrate sequences. This technique allows quantification of DNA polymerase activity using different templates and under different conditions with similar sensitivity to the radioactive assay. ACKNOWLEDGMENTS Research supported by CNPq-Brazil and PRONEX. S.M.R. Teixeira and C.R. Machado were supported, in part, by an International Research Scholarship Grant from the Howard Hughes Medical Institute. We are grateful to Kátia Barroso Gonçalves and Neuza Antunes Rodrigues for technical support. REFERENCES Braithwaite DK and Ito J (1993). Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 21: 787-802. Filee J, Forterre P, Sen-Lin T and Laurent J (2002). Evolution of DNA polymerase families: evidences for multiple gene exchange between cellular and viral proteins. J. Mol. Evol. 54: 763-773. Horhota A, Zou K, Ichida JK, Yu B, et al. (2005). Kinetic analysis of an efficient DNA-dependent TNA polymerase. J. Am. Chem. Soc. 127: 7427-7434. Lestienne P, Pourquier P and Bonnet J (2003). Elongation of oligonucleotide primers forming a triple helix on double-stranded DNA templates by purified DNA polymerases. Biochem. Biophys. Res. Commun. 311: 380-385. Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, et al. (2006). Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr. Biol. 16: 202-207. Pelletier H, Sawaya MR, Kumar A, Wilson SH, et al. (1994). Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science 264: 1891-1903. Prober JM, Trainor GL, Dam RJ, Hobbs FW, et al. (1987). A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science 238: 336-341. Roettger MP, Fiala KA, Sompalli S, Dong Y, et al. (2004). Pre-steady-state kinetic studies of the fidelity of human DNA polymerase mu. Biochemistry 43: 13827-13838. Smith LM, Kaiser RJ, Sanders JZ and Hood LE (1987). The synthesis and use of fluorescent oligonucleotides in DNA sequence analysis. Methods Enzymol. 155: 260-301. Tippin B, Pham P and Goodman MF (2004). Error-prone replication for better or worse. Trends Microbiol. 12: 288-295. Tuske S, Singh K, Kaushik N and Modak MJ (2000). The J-helix of Escherichia coli DNA polymerase I (Klenow fragment) regulates polymerase and 3'- 5'-exonuclease functions. J. Biol. Chem. 275: 23759-23768. Yagi Y, Ogawara D, Iwai S, Hanaoka F, et al. (2005). DNA polymerases eta and kappa are responsible for error-free translesion DNA synthesis activity over a cis-syn thymine dimer in Xenopus laevis oocyte extracts. DNA Repair 4: 1252-1269. Zhang X, Lee I and Berdis AJ (2005). A potential chemotherapeutic strategy for the selective inhibition of promutagenic DNA synthesis by nonnatural nucleotides. Biochemistry 44: 13111-13121. |
|