![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
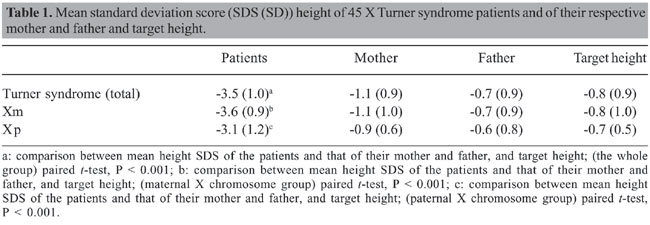
ABSTRACT. Thirty-seven 45 X Turner syndrome patients with confirmed peripheral blood lymphocyte karyotype were initially selected to determine the origin of the retained X chromosome and to correlate it with their parents’ stature. Blood samples were available in 25 families. The parental origin of the X chromosome was determined in 24 informative families through the analysis of the exon 1 - CAG repeat variation of the androgen receptor gene. In 70.8% of the cases, the retained X chromosome was maternal in origin and 29.2% was paternal. When we classified the patients according to maternal (Xm) or paternal (Xp) X chromosome, there was a positive correlation between patients’ and maternal heights only in the Xm group. There was no correlation with paternal height in either group, and a significant correlation with target height was only observed in the Xm group. In conclusion, maternal height is the best variable correlating with the height of 45 X Turner syndrome patients who retain the maternal X chromosome, suggesting a strong influence of genes located on the maternal X chromosome on stature. Key words: Turner syndrome, X chromosome, Parental origin, Short stature INTRODUCTION Height remains an important psychosocial determinant, and short stature represents one of the most frequent complaints in pediatrics and in pediatric endocrinology clinics. Although not always is secondary to a pathological condition, height has been defined as being under normal limits when below -2 standard deviation (SD) for age and sex (Longui, 1998). A positive correlation has been found between the height of a given individual and the height of his/her parents, and the data suggest a combined influence of multiple genes in the determination of final height (Reiter and Rosenfeld, 1998). Among these genes, some have been localized on the short arm of the X chromosome, especially the short stature homeobox-containing gene - SHOX (Rao et al., 1997). Familial target height can be calculated as suggested by Tanner et al. (1970). Nonetheless, this method of target height determination is not useful if the heights of the father and mother are discordant (parental height difference greater than 2 SD). In such cases, each individual seems to randomly follow the SD stature from the mother or father. This phenomenon could, in part, be related to a predominant gene expression on the X chromosome, representing the skewed expression of a given parental X chromosome. Patients with 45 X Turner syndrome (TS) have only one X chromosome of paternal or maternal origin. This represents a unique clinical model in which the influence of X chromosome genes related to their parental origin is manifested. Previous studies have attempted to correlate the stature of TS patients with that of their parents. The results have been controversial, with this correlation being significant for both maternal or paternal stature or, in some cases, only with the target height (Salerno and Job 1987; Massa et al., 1990; Cohen et al., 1995). In some cohorts, contradictory findings can be dependent on the inclusion of different karyotypes. The aim of the present study was to determine the influence of parental origin of the X chromosome on the stature of patients with 45 X TS. PATIENTS AND METHODS Thirty-seven 45 X TS patients with confirmed peripheral blood lymphocyte karyotype were initially selected, after an informed consent form was signed by the parents or guardians and the study approved by the ethics committee of the institution. Blood samples for DNA extraction were available from the patients and their parents in 25 families. The clinical data obtained were gestational age, birth weight and height, weight and height before treatment with growth hormone, and parental height. Weight and height were expressed as standard deviation scores (SDS) according to NCHS-2000 reference data (Centers for Disease Control and Prevention, 2006). Target height was calculated according to the formula described by Tanner et al. (1970) and also expressed as SDS. Patients or parents with systemic chronic diseases or endocrine or other genetic abnormalities with potential influence on stature were excluded. Patients with karyotypes different from 45 X, even low frequency mosaicism (Longui et al., 2002), were also excluded. Molecular analysis The parental origin of the X chromosome was determined taking advantage of the very high frequency of polymorphism observed in the exon 1 - CAG repeats of the androgen receptor (AR) gene (HUMARA gene, OMIM: 313700; located in the Xq11-12 region). DNA samples were submitted to PCR amplification, employing primers targeted to the exon 1 of the AR gene. The sense primer was labeled with 6-FAM (6-carboxy-fluorescein) and synthesized as follows: 5' 6-FAM - GGG TAA GGG AAG TAG GTG GAA 3' (Invitrogen Corporation, Carlsbad, CA, USA). The non-labeled antisense primer was: 5' ACT GCG GCT GTG AAG GTT 3'. PCR included 5 pmol of the labeled sense primer, 20 pmol of non-labeled antisense primer, 200 µmol of each dNTP, 10X buffer, 0.1 U Taq DNA polymerase (GeneAmp - PCR reagent kit with AmpliTaq DNA polymerase - Perkin Elmer, Branchburg, NJ, USA), 50 mM MgCl2, and ddH2O to obtain a final volume of 50 µL. PCR was run in a GeneAmp PCR System 9700 (Applied Biosystem, Foster City, CA, USA) thermocycler under the following conditions: initial denaturing step at 94°C for 3 min, followed by 30 cycles of denaturation, annealing and extension (0.5, 1 and 1 min, respectively). A final extension at 72°C for 7 min was also performed. The PCR product obtained (395-bp fragment) was then submitted to capillary gel electrophoresis in an ABI PRISM 310 automated analyzer (Applied Biosystems) under the following conditions: temperature of 60°C, 7-9 µA° for 28 min for each sample. The results were analyzed employing GeneScan software which is able to determine the relative size of the amplified fragment, where the latter variable depends on the number of CAG repeats present in the exon 1 of the AR gene. Using this method, the parental origin of the X chromosome can be determined, comparing the patient’s allele size to the alleles observed in the respective mother and father. Statistical analysis Statistical analyses were performed employing the SPSS for Windows software, version 10.0.1 (SPSS Inc., Chicago, IL, USA). Student t-test was used to compare variables between the two groups of patients, established according to maternal or paternal origin of the X chromosome. Spearman’s correlation coefficient between the patient’s height and the mother’s or father’s stature was established for each study group. The frequencies of other clinical variables, such as cardiac, renal and orthopedic diseases and Hashimoto’s thyroiditis were also compared between the two groups. RESULTS Of the 25 families in which DNA samples were available, only one was considered non-informative because the maternal and paternal alleles were found to be the same size. Therefore, the results of this study are based on the remaining 24 informative families. All TS patients had short stature compared to the normal population or target height. Mean SDS height (SD) was -3.5 (1.0). At birth, mean (SD) weight and height were 2610 (522) g and 46 (2.4) cm, respectively. After correcting the gestational age, 3/22 (13.6%) and 13/19 (68.4%), patients were found to be below -2 SDS for birth weight and height, respectively. The mean (SD) for maternal height SDS was observed to be -1.1 (0.9), -0.7 (0.9) for paternal height SDS and -0.8 (0.9) for target height SDS. A significant decrease was seen in patient height when compared with maternal, paternal or target height (Table 1).
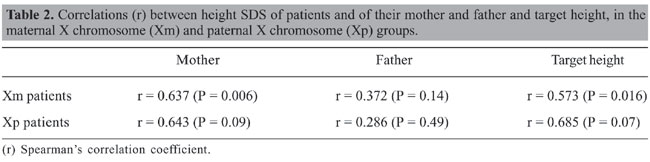
Maternal origin of the X chromosome was detected in 17/24 TS patients (70.8%), and paternal origin detected in the remaining 7/24 cases (29.2%). When the patients were divided into two groups, according to maternal (Xm) or paternal (Xp) origin of the X chromosome, there were no significant differences between the two groups regarding chronological age, birth weight and height, and SD scores for maternal, paternal or target height. As observed for the whole group, the patients’ height SDS of the Xm or Xp groups also differed significantly from maternal, paternal or target height (Table 1). Spearman’s correlation coefficients between the height of TS patients and their parents’ or target height are presented in Table 2. In the Xm group, there was a positive correlation between the patients’ and respective maternal height, but not with paternal height. No similar correlation was observed in patients with Xp origin. Neither of the groups showed a significant correlation between patient height and respective paternal height. A significant correlation with target height was observed only in the Xm group, although a tendency toward a significant difference was also observed in the Xp group.
There were no significant differences between the patients of the Xm and Xp groups in the frequency of cardiac, renal and orthopedic abnormalities or Hashimoto’s thyroiditis. The frequencies of low-birth weight and height SDS were also similar in both groups. DISCUSSION An increasing number of genes have been identified as being involved in the modulation of growth, and after reports of SHOX mutations, the influence of other X chromosome genes have been studied (Page et al., 1987; Luoh et al., 1997). On the other hand, there is no general consensus that sex chromosome-related genes are the major determinants of final height, and other autosomal genes have been implicated in growth modulation (Arinami et al., 1999; Deng et al., 2002; Wiltshire et al., 2002). The origin of the X chromosome and, consequently, the stature of the respective parent can be one of the factors determining the final height of an individual. In the cohort examined in the present study, which included only 45 X TS patients, around 70% received the X chromosome from the mother (maternal monosomy); similar results were observed by others (Mathur et al., 1991; Lorda-Sanchez et al., 1992; Chu et al., 1994; Jacobs et al., 1990, 1997; Monroy et al., 2002; Uematsu et al., 2002). In the series described by Loughlin et al. (1991), all patients preserved the Xm chromosome, although in the cases reported by Tsezou et al. (1999), the X chromosome was of maternal origin in only 7/16 (43.7%). In the majority of previously reported studies, more laborious and less sensitive assays, such as Southern blotting or restriction fragment length polymorphism analysis, were employed. More recently, Monroy et al. (2002) used a PCR-amplified labeled fragment to detect parental origin of the X chromosome, but several polymorphic markers were needed for accurate identification. In our study employing GeneScan methodology aimed at the characterization of the AR gene polymorphism, the origin of the X chromosome could not be defined in only one of the 25 families, demonstrating the high sensitivity and precision of this method. Additionally, this method has the advantage of being rapid and sensitive to rule out the presence of low-frequency X0/XX mosaics, even when employing only one polymorphic marker of the X chromosome. We also confirm that there is a significant correlation between TS patients’ height and maternal height in cases with Xm origin. There was no correlation with the paternal stature for both Xm and Xp patients. Similar results were also found in the TS patients described by Chu et al. (1994), despite the fact that the authors did not classify the patients according to their karyotype. Others found a correlation with maternal stature in patients with either Xm or Xp origin (Tsezou et al., 1999). However, the karyotype was also not considered in the latter study, and the patients’ SDS were calculated employing the reference data of patients with TS, which could overestimate the patient’s SDS. Although the mean final height of patients with TS tends to be in a narrow range, a substantial individual variability can be observed in the TS patient’s height. This can be explained, at least in part, by the frequently observed variability in parental height SDS. The conclusion of the present study evaluating the stature of patients with 45 X TS is that the best correlation is observed in TS patients that retain the Xm chromosome and their maternal height, suggesting the strong influence of genes located on the maternal X chromosome in the modulation of growth and the determination of final height. ACKNOWLEDGMENTS We are grateful to the Support Center for Scientific Publications of Santa Casa de São Paulo, Faculty of Medical Sciences, São Paulo, SP, Brazil, for editorial assistance. REFERENCES Arinami T, Iijima Y, Yamakawa-Kobayashi K, Ishiguro H, et al. (1999). Supportive evidence for contribution of the dopamine D2 receptor gene to heritability of stature: linkage and association studies. Ann. Hum. Genet. 63 (Pt 2): 147-151. Centers for Disease Control and Prevention (2006). www.cdc.gov/growthcharts. Chu CE, Donaldson MD, Kelnar CJ, Smail PJ, et al. (1994). Possible role of imprinting in the Turner phenotype. J. Med. Genet. 31: 840-842. Cohen A, Kauli R, Pertzelan A, Lavagetto A, et al. (1995). Final height of girls with Turner’s syndrome: correlation with karyotype and parental height. Acta Paediatr. 84: 550-554. Deng HW, Xu FH, Liu YZ, Shen H, et al. (2002). A whole-genome linkage scan suggests several genomic regions potentially containing QTLs underlying the variation of stature. Am. J. Med. Genet. 113: 29-39. Jacobs PA, Betts PR, Cockwell AE, Crolla JA, et al. (1990). A cytogenetic and molecular reappraisal of a series of patients with Turner’s syndrome. Ann. Hum. Genet. 54 (Pt 3): 209-223. Jacobs P, Dalton P, James R, Mosse K, et al. (1997). Turner syndrome: a cytogenetic and molecular study. Ann. Hum. Genet. 61 (Pt 6): 471-483. Longui C (1998). Crescimento deficiente. In: Endocrinologia para o pediatra (Monte O, Longui C and Calliari LEP, eds.). Atheneu, São Paulo, 11-18. Longui CA, Rocha MN, Martinho LC, Gomes GG, et al. (2002). Molecular detection of XO - Turner syndrome. Genet. Mol. Res. 1: 266-270. Lorda-Sanchez I, Binkert F, Maechler M and Schinzel A (1992). Molecular study of 45,X conceptuses: correlation with clinical findings. Am. J. Med. Genet. 42: 487-490. Loughlin SA, Redha A, McIver J, Boyd E, et al. (1991). Analysis of the origin of Turner’s syndrome using polymorphic DNA probes. J. Med. Genet. 28: 156-158. Luoh SW, Bain PA, Polakiewicz RD, Goodheart ML, et al. (1997). Zfx mutation results in small animal size and reduced germ cell number in male and female mice. Development 124: 2275-2284. Massa G, Vanderschueren-Lodeweyckx M and Malvaux P (1990). Linear growth in patients with Turner syndrome: influence of spontaneous puberty and parental height. Eur. J. Pediatr. 149: 246-250. Mathur A, Stekol L, Schatz D, MacLaren NK, et al. (1991). The parental origin of the single X chromosome in Turner syndrome: lack of correlation with parental age or clinical phenotype. Am. J. Hum. Genet. 48: 682-686. Monroy N, Lopez M, Cervantes A, Garcia-Cruz D, et al. (2002). Microsatellite analysis in Turner syndrome: parental origin of X chromosomes and possible mechanism of formation of abnormal chromosomes. Am. J. Med. Genet. 107: 181-189. Page DC, Mosher R, Simpson EM, Fisher EM, et al. (1987). The sex-determining region of the human Y chromosome encodes a finger protein. Cell 51: 1091-1104. Rao E, Weiss B, Fukami M, Rump A, et al. (1997). Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 16: 54-63. Reiter OE and Rosenfeld RG (1998). Normal and aberrant growth. In: Williams textbook of endocrinology (Wilson JD, Foster DW, Kronenberg HM and Larsen PR, eds.). W.B. Saunders Company, Philadelphia, 1427-1507. Salerno MC and Job JC (1987). [Height in Turner’s syndrome: correlation with parents’ height] La taille dans le syndrome de Turner: corrélations avec la taille des parents. Arch. Fr. Pediatr. 44: 863-865. Tanner JM, Goldstein H and Whitehouse RH (1970). Standards for children’s height at ages 2-9 years allowing for heights of parents. Arch. Dis. Child. 45: 755-762. Tsezou A, Hadjiathanasiou C, Gourgiotis D, Galla A, et al. (1999). Molecular genetics of Turner syndrome: correlation with clinical phenotype and response to growth hormone therapy. Clin. Genet. 56: 441-446. Uematsu A, Yorifuji T, Muroi J, Kawai M, et al. (2002). Parental origin of normal X chromosomes in Turner syndrome patients with various karyotypes: implications for the mechanism leading to generation of a 45,X karyotype. Am. J. Med. Genet. 111: 134-139. Wiltshire S, Frayling TM, Hattersley AT, Hitman GA, et al. (2002). Evidence for linkage of stature to chromosome 3p26 in a large U.K. family data set ascertained for type 2 diabetes. Am. J. Hum. Genet. 70: 543-546. |
|