![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
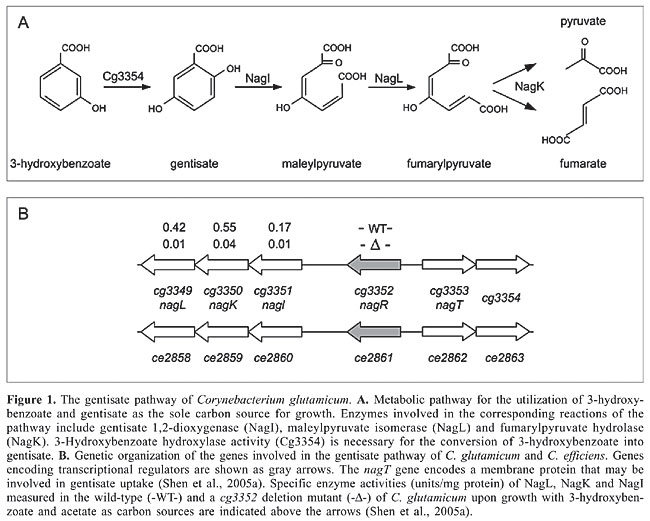
ABSTRACT. Corynebacterium glutamicum is a gram-positive soil microorganism able to utilize a large variety of aromatic compounds as the sole carbon source. The corresponding catabolic routes are associated with multiple ring-fission dioxygenases and among other channeling reactions, include the gentisate pathway, the protocatechuate and catechol branches of the b-ketoadipate pathway and two potential hydroxyquinol pathways. Genes encoding the enzymatic machinery for the bioconversion of aromatic compounds are organized in several clusters in the C. glutamicum genome. Expression of the gene clusters is under specific transcriptional control, apparently including eight DNA-binding proteins belonging to the AraC, IclR, LuxR, PadR, and TetR families of transcriptional regulators. Expression of the gentisate pathway involved in the utilization of 3-hydroxybenzoate and gentisate is positively regulated by an IclR-type activator. The metabolic channeling of ferulate, vanillin and vanillate into the protocatechuate branch of the b-ketoadipate pathway is controlled by a PadR-like repressor. Regulatory proteins of the IclR and LuxR families participate in transcriptional regulation of the branches of the b-ketoadipate pathway that are involved in the utilization of benzoate, 4-hydroxybenzoate and protocatechuate. The channeling of phenol into this pathway may be under positive transcriptional control by an AraC-type activator. One of the potential hydroxyquinol pathways of C. glutamicum is apparently repressed by a TetR-type regulator. This global analysis revealed that transcriptional regulation of aromatic compound utilization is mainly controlled by single regulatory proteins sensing the presence of aromatic compounds, thus representing single input motifs within the transcriptional regulatory network of C. glutamicum. Key words: Corynebacterium glutamicum, Corynebacterium efficiens, Gentisate pathway, b-ketoadipate pathway, Hydroxyquinol pathway, Transcriptional regulation INTRODUCTION Microorganisms constantly have to adapt to changing environmental conditions in order to maintain their functional homeostasis and to handle different types of environmental and cellular stresses (Matic et al., 2004). Therefore, they sense alterations within their surrounding and apply complex molecular strategies involving transcriptional regulation of gene expression to overcome unfavorable conditions. DNA-binding transcriptional regulators are the most important components of these regulatory systems since they specifically detect the presence of effector molecules to exert their regulatory role in gene expression (Madan Babu and Teichmann, 2003a; Martinez-Antonio et al., 2006). Depending on the environmental conditions, only certain fractions of the genome of a bacterial cell are expressed, according to the behavior of transcriptional regulators within the complete network of regulatory interactions (Resendis-Antonio et al., 2005). While some regulatory proteins are only responsible for the transcriptional control of single genes or gene clusters, others organize the activation or repression of numerous target genes (Teichmann and Babu, 2004). Nowadays, knowledge of the whole genome sequence of a microorganism provides the possibility to determine the total set of encoded DNA-binding transcriptional regulators (Perez-Rueda and Collado-Vides, 2000; Guedon et al., 2002; Brune et al., 2005; Moreno-Campuzano et al., 2006) and to apply bioinformatics and post-genomic techniques, such as DNA microarray hybridizations, to reconstruct the global connectivity of bacterial regulatory networks (Herrgard et al., 2004). Corynebacterium glutamicum is a gram-positive soil bacterium that is traditionally applied in biotechnological production processes, particularly in the fermentative production of amino acids (Hermann, 2003). The complete genome sequence of the wild-type strain C. glutamicum ATCC 13032 provided the basis to identify and analyze the repertoire of transcriptional regulators in this species (Kalinowski et al., 2003; Brune et al., 2005). The total set of DNA-binding transcriptional regulators of C. glutamicum was determined to be 127 proteins that can be further associated with one of five well-defined and functionally distinct regulatory modules with respect to the physiological role of their target genes (Brune et al., 2005; Baumbach et al., 2006). As a free-living soil bacterium, C. glutamicum is exposed to permanent variations in the supply of nutrients specified by its natural habitat so that a fast adaptation to changing environmental conditions is of great importance to allow optimal growth and reproduction. The high demand for flexibility in the consumption of different carbon and energy sources may be the reason why a large proportion of regulatory proteins can be associated with transcriptional control of bacterial carbohydrate metabolism (Guedon et al., 2002; Brune et al., 2005; Resendis-Antonio et al., 2005). Besides being able to degrade sugars, C. glutamicum also shows the ability to utilize a large variety of aromatic compounds for growth (Shen and Liu, 2005; Shen et al., 2005a,b; Merkens et al., 2005). Aromatic compounds, such as the complex aromatic polymer lignin, are natural components of the environment of soil bacteria and represent an alternative source to sugars for carbon and energy (Harwood and Parales, 1996). The presence of oxygen is a precondition for a fast and effective assimilation of aromatic compounds, although anaerobic decomposition by some bacteria is also possible. The catabolic reactions take place in two principle steps comprising the preparation of enzymatic ring cleavage, termed the peripheral pathway, and the ring-fission itself (Diaz, 2004). To prepare aromatic compounds for ring cleavage, a variety of chemical modifications involving mono- or dioxygenation are carried out. The ring modification reactions of the peripheral pathways result in the formation of dihydroxylated benzene derivatives, such as gentisate, protocatechuate and catechol. These intermediate compounds are the substrates of specific ring-fission enzymes that integrate oxygen to open the aromatic ring in the second phase of degradation (Harwood and Parales, 1996). Fission takes place either between two adjacent hydroxyl groups (ortho cleavage) or proximal to one of the two hydroxyl groups (meta cleavage) of the aromatic compound. A third ring cleavage pathway processes aromatic compounds, such as gentisate, whose two hydroxyl groups on the aromatic ring are para to each other (Harwood and Parales, 1996). In principle, structurally diverse aromatic compounds are degraded through different peripheral pathways to only few intermediates that are further channeled via common reactions into the central carbohydrate metabolism of the bacterial cell (Diaz, 2004). In this review, we summarize the current knowledge of the ability of C. glutamicum to utilize various aromatic compounds as the sole source of carbon and energy for growth. We will present the structure of the different catabolic pathways of aromatic compounds identified in the C. glutamicum genome sequence and the specific ring-fission dioxygenases they involve. Furthermore, we will connect these catabolic pathways to transcriptional regulation carried out by different types of regulatory proteins. TRANSCRIPTIONAL REGULATION OF THE GENTISATE PATHWAY OF CORYNEBACTERIUM GLUTAMICUM C. glutamicum can utilize 3-hydroxybenzoate and gentisate as the sole carbon source for growth (Shen et al., 2005a). Gentisate is degraded to pyruvate and fumarate, whereas 3-hydroxybenzoate can be converted into gentisate by 3-hydroxybenzoate 6-hydroxylase activity (Figure 1A). The relevant ring-cleavage activity of this pathway is provided by gentisate 1,2-dioxygenase (Harwood and Parales, 1996). A cluster of six genes (cg3349 to cg3354) was recently shown to encode the enzymes involved in the gentisate pathway of C. glutamicum (Figure 1B; Shen et al., 2005a). A homologous gene cluster (ce2858 to ce2863) is present in the genome of C. efficiens (Figure 1B; Nishio et al., 2003). When C. glutamicum cells were cultivated with 3-hydroxybenzoate and gentisate as the sole carbon source, gentisate 1,2-dioxygenase activity was detected, suggesting that both aromatic compounds are assimilated by the gentisate pathway (Shen et al., 2005a). The respective enzymatic functions were deduced from enzyme assays with purified proteins and from genetic studies using defined deletion mutants of C. glutamicum, providing evidence that cg3349 (nagL), cg3350 (nagK) and cg3351 (nagI) encode maleylpyruvate isomerase, fumarylpyruvate hydrolase and gentisate 1,2-dioxygenase, respectively (Hintner et al., 2004; Shen et al., 2005a). The Cg3349 protein represents a new type of maleylpyruvate isomerase since it is independent of glutathione and requires mycothiol as co-factor (Shen et al., 2005a; Feng et al., 2006). Furthermore, deletion of the cg3353 (nagT) gene encoding a transport protein resulted in the loss of the ability to utilize gentisate, but did not affect the growth of C. glutamicum with 3-hydroxybenzoate, suggesting that cg3353 codes for a gentisate transporter (Shen et al., 2005a). This result was substantiated very recently by the heterologous expression and analysis of a Cg3353-GFP fusion protein in Ralstonia sp strain U2 (Xu et al., 2006). On the other hand, deletion of cg3354 negatively affected the growth of C. glutamicum with 3-hydroxybenzoate. Escherichia coli cells expressing the Cg3354 protein converted 3-hydroxybenzoate into gentisate, indicating that the protein has 3-hydroxybenzoate hydroxylase activity (Figure 1A).
A regulatory gene encoding a transcriptional regulator of the IclR family (Table 1; Brune et al., 2005) is located upstream of the gentisate 1,2-dioxygenase gene (Figure 1B). Deletion of the cg3352 (nagR) gene resulted in significant loss of the enzyme activities encoded by cg3351, cg3350 and cg3349 (Figure 1B) and of the ability to grow with 3-hydroxybenzoate and gentisate as the sole carbon source (Shen et al., 2005a). These data demonstrated that at least the genes cg3349 to cg3351 are under positive transcriptional control and that cg3352 encodes an activator protein of the gentisate pathway (Table 1). Activation of expression of the gentisate pathway thus requires both a functional Cg3352 (NagR) protein and the presence of either gentisate or 3-hydroxybenzoate that could be converted into gentisate by hydroxylation.
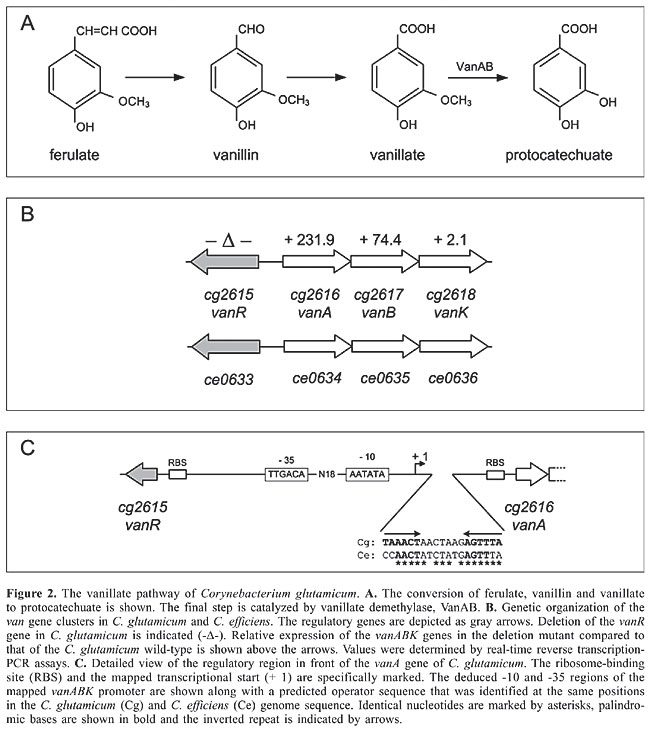
TRANSCRIPTIONAL REGULATION OF THE VANILLATE PATHWAY OF CORYNEBACTERIUM GLUTAMICUM Microbiological assays revealed that C. glutamicum is able to grow on ferulate, vanillin and vanillate as the sole carbon source (Merkens et al., 2005; Shen et al., 2005b). Bioconversion of ferulate, vanillin and vanillate leads to protocatechuate, thus channeling these aromatic compounds into the protocatechuate branch of the b-ketoadipate pathway (Figure 2A). Thereby, enzymatic conversion of vanillate into protocatechuate is catalyzed by vanillate demethylase (Mason and Cammack, 1992). A cluster of four genes (cg2615 to cg2618) has been associated with vanillate metabolism in C. glutamicum (Merkens et al., 2005), including the vanA, vanB and vanK genes and the divergently transcribed vanR gene (Figure 2B). A similar gene cluster (ce0633 to ce0636) has been detected in the genome sequence of C. efficiens (Figure 2B; Nishio et al., 2003). The deduced proteins of the vanAB genes of C. glutamicum showed significant similarity to vanillate demethylase subunits A and B from different microorganisms (Kalinowski et al., 2003). A C. glutamicum mutant carrying a disrupted vanA gene was unable to grow on ferulate and vanillate, whereas growth on protocatechuate was not impaired (Merkens et al., 2005). The vanK gene has been suggested to encode a protocatechuate transporter since the metabolism of ferulate and vanillate was not impaired in a vanK disruption mutant, while growth on protocatechuate was biphasic. Further growth experiments showed that glucose and vanillate were co-metabolized by C. glutamicum, revealing no indication for the participation of glucose-mediated carbon catabolite response in the expression of the van genes. Induction of expression of the vanABK genes was observed with ferulate and vanillate, while protocatechuate had no positive effect on vanA gene expression (Merkens et al., 2005). Reverse transcription-PCR assays revealed a common transcript of the vanABK cluster, suggesting that the genes are transcriptionally organized as an operon. The common transcript of vanABK starts 59 nucleotides upstream of the vanA start codon as has been determined by 5' RACE experiments, resulting in the mapping of a potential promoter region (Figure 2C).
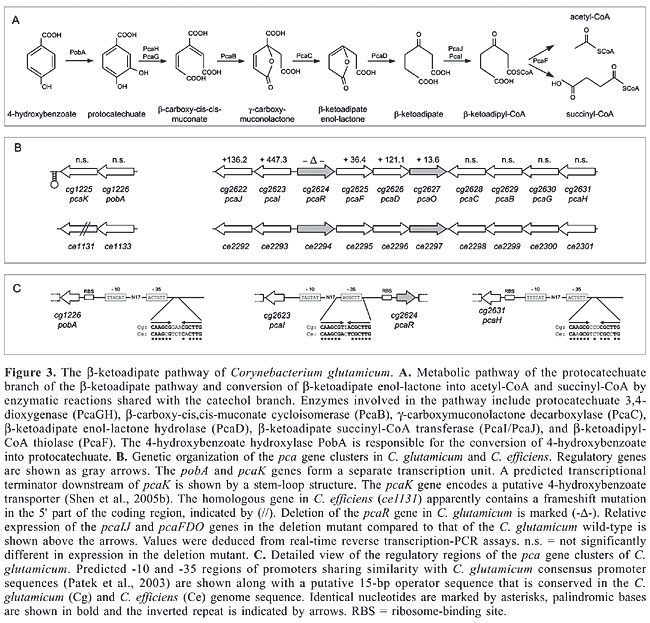
The vanR gene (cg2615) codes for a DNA-binding protein that has been classified in the PadR family of transcriptional regulators (Table 1; Brune et al., 2005). This protein family includes, for instance, functionally characterized members that are involved in the negative transcriptional regulation of phenolic acid metabolism (Bateman et al., 2002). The regulatory role of vanR in vanABK gene expression was deduced from a defined deletion mutant and comparison of expression of the vanABK genes with that of the C. glutamicum wild-type strain. Deletion of vanR resulted in enhanced expression of the vanABK genes, demonstrating that VanR control of vanABK expression is exerted negatively at the level of transcription (Figure 2B). A region of short dyad symmetry was identified downstream of the mapped vanABK promoter, indicating a putative operator sequence of a repressor protein (Figure 2C; Madan Babu and Teichmann, 2003b). A similar stretch of DNA was identified in the regulatory region of the vanABK genes of C. efficiens. Likewise, predicted operator sequences of related PadR transcriptional regulators identified in Pediococcus pentosaceus (Barthelmebs et al., 2000) and Lactobacillus plantarum (Gury et al., 2004) are apparently organized in a similar manner, consisting of a short stretch of dyad symmetry along with a non-palindromic center. TRANSCRIPTIONAL REGULATION OF THE b-KETOADIPATE PATHWAY OF CORYNEBACTERIUM GLUTAMICUM The spectrum of aromatic compound utilization by C. glutamicum also includes 4-hydroxybenzoate, protocatechuate and p-cresol (Shen and Liu, 2005). Assimilation of 4-hydroxybenzoate and protocatechuate apparently occurs by the b-ketoadipate pathway, consisting of the protocatechuate and catechol branches that converge at b-ketoadipate enol-lactone and a central pathway of three additional steps leading to the final products acetyl-CoA and succinyl-CoA (Figure 3A; Harwood and Parales, 1996). p-Cresol may be channeled into the b-ketoadipate pathway by conversion into 4-hydroxybenzoate (Shen et al., 2005b). The key activity of the protocatechuate branch of the b-ketoadipate pathway is represented by protocatechuate 3,4-dioxygenase which is responsible for aromatic ring cleavage. Cells grown in the presence of 4-hydroxybenzoate, protocatechuate and p-cresol exhibited significant protocatechuate 3,4-dioxygenase activity (Shen and Liu, 2005), indicating that expression of the protocatechuate branch of the b-ketoadipate pathway is strictly regulated and dependent on the corresponding substrates as inducers. Furthermore, cultivation of C. glutamicum in the presence of protocatechuate and glucose revealed no indication for a superior glucose-mediated carbon catabolite control of the b-ketoadipate pathway (Merkens et al., 2005). The protocatechuate branch of the b-ketoadipate pathway is encoded by a supraoperonic cluster of ten genes (cg2622 to cg2631 and ce2292 to ce2301) in the genomes of C. glutamicum and C. efficiens (Figure 3B; Kalinowski et al., 2003; Nishio et al., 2003). The subunits of protocatechuate 3,4-dioxygenase are obviously encoded by cg2630 (pcaG) and cg2631 (pcaH) since deletion of the pcaGH genes in the C. glutamicum genome resulted in a mutant strain that had lost protocatechuate 3,4-dioxygenase activity and that was unable to utilize 4-hydroxybenzoate, protocatechuate and p-cresol as the sole carbon source for growth (Shen and Liu, 2005). The physiological function of additional genes involved in the protocatechuate branch of the b-ketoadipate pathway was deduced from sequence similarity searches (Figure 3A; Shen et al., 2005b). 4-Hydroxybenzoate hydroxylase activity, necessary for the conversion of 4-hydroxybenzoate into protocatechuate, is most likely encoded by the pobA (cg1226) gene which is located in a separate transcription unit along with the pcaK (cg1225) gene encoding a putative 4-hydroxybenzoate transporter (Figure 3B; Shen et al., 2005b).
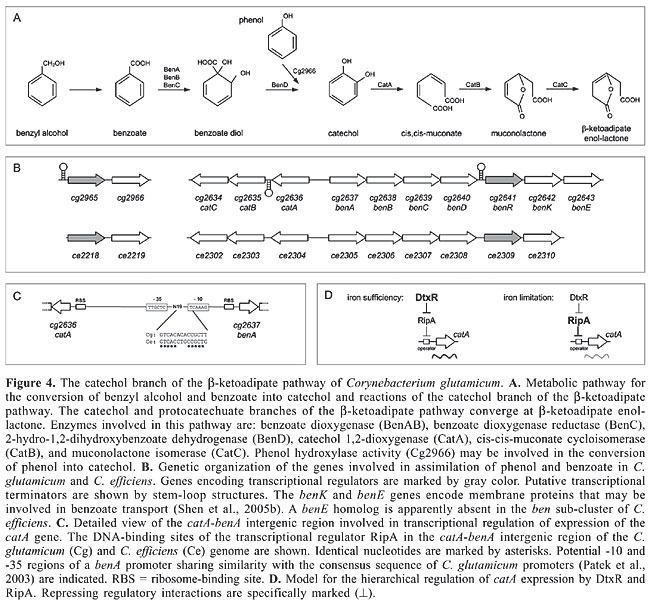
Two genes encoding transcriptional regulators are part of the pca gene clusters in C. glutamicum and C. efficiens (Figure 3B). The pcaR gene encodes a transcriptional regulator of the IclR family and pcaO codes for a transcriptional activator that was grouped into the LuxR protein family (Table 1; Brune et al., 2005). The role of the PcaO protein in the regulation of the transcription of the pca genes is currently unknown, whereas the respective regulatory function of the PcaR protein can be deduced from sequence similarity searches. Accordingly, it is likely that PcaR exerts its regulatory role in the expression of the pca genes by interacting with 15-bp operator sequences that are located in the pcaI-pcaR intergenic region and upstream of pcaH and pobA (Figure 3C), indicating that the expression of each sub-cluster is under direct transcriptional control by PcaR. Similar partially palindromic sequences are apparently conserved at precisely the same positions in the pca gene clusters of C. efficiens (Figure 3C). The consensus sequence of the 15-bp operators detected in the C. glutamicum genome is represented by the partially palindromic motif 5'-GTTCGC-N3-GCGAAC-3'. It is worthwhile to note that regulatory proteins of the b-ketoadipate pathway in other bacteria use similar DNA-binding sites for transcriptional regulation. This includes the transcriptional activator PcaR from Pseudomonas putida (Romero-Steiner et al., 1994; Guo and Houghton, 1999) and the activators PobR and PcaU from Acinetobacter calcoaceticus (DiMarco and Ornston, 1994; Gerischer et al., 1998), indicating that transcriptional control by homologous proteins with the same physiological function is associated with highly similar operator sequences. However, the PcaR protein of C. glutamicum is a member of the IclR family of transcriptional regulators that can also act as repressors of gene expression (Molina-Henares et al., 2006). When inspecting the very short pcaI-pcaR intergenic region of C. glutamicum, which is only 64 bp in length, it is apparent that the 15-bp operator of PcaR overlaps the potential promoter of pcaI and is located directly adjacent to the ribosome-binding site of the pcaR gene (Figure 3C). This genetic organization of the intergenic region is more consistent with a negative regulatory interaction of the PcaR protein (Madan Babu and Teichmann, 2003b). Accordingly, a defined deletion of the pcaR gene of C. glutamicum resulted in elevated levels of expression of the pcaIJ and pcaFDO genes as deduced from real-time reverse transcription-PCR assays (Figure 3B). On the other hand, expression of the pcaHGBC genes and of the pobA-pcaK transcription unit was not significantly affected by the pcaR deletion. This data supported the presumption that PcaR acts as a repressor of gene expression, including the regulatory gene pcaO (Table 1). This type of hierarchical regulation of pca gene expression is reasonable when considering that the b-ketoadipate pathway consists of two specific parallel routes and a common terminal portion (Harwood and Parales, 1996). The pcaIJ and pcaFD genes are involved in the common part of the b-ketoadipate pathway and expression is negatively regulated only by the PcaR protein (Figure 3A, B). On the other hand, the pcaHGBC and pobA-pcaK genes, belonging to the protocatechuate branch of the b-ketoadipate pathway, are also under negative transcriptional control by PcaR, but deletion of the pcaR gene alone did not result in elevated expression of these genes. This could indicate that a second regulator is involved in triggering the transcriptional response of the specific part of the b-ketoadipate pathway depending on the aromatic compounds present in the environment. This additional regulatory role may be attributed to the LuxR-type transcriptional activator PcaO (Table 1; Brune et al., 2005), which in this way allows a decoupling of the regulation of genes belonging to the specific part of the b-ketoadipate pathway from the more flexible derepression of the common pathway necessary for the utilization of a larger variety of aromatic compounds. However, the precise role of the PcaO regulator in controlling the expression of the protocatechuate branch of the b-ketoadipate pathway remains to be elucidated. TRANSCRIPTIONAL REGULATION OF THE CATECHOL BRANCH OF THE b-KETOADIPATE PATHWAY OF CORYNEBACTERIUM GLUTAMICUM C. glutamicum can also use benzyl alcohol, benzoate and phenol as the sole carbon and energy source for growth (Shen et al., 2004, 2005b). Benzoate and phenol are representative compounds that are degraded by the catechol branch of the b-ketoadipate pathway (Figure 4A; Harwood and Parales, 1996). Enzyme assays revealed that C. glutamicum cells grown in the presence of these aromatic compounds showed catechol 1,2-dioxygenase activity (Shen et al., 2004, 2005b), indicating a tight regulation of the respective catabolic pathway. The genes involved in the bioconversion of benzoate to b-ketoadipate enol-lactone are organized in a single cluster in the genomes of C. glutamicum and C. efficiens (Kalinowski et al., 2003; Nishio et al., 2003), comprising the divergently oriented ben and cat sub-clusters (Figure 4B). Benzoate degradation requires the ben genes for converting benzoate into catechol and the cat genes for degrading the resulting catechol. The physiological role of the respective proteins has been deduced from sequence similarity searches with characterized counterparts from gram-positive and gram-negative bacteria (Figure 4A; Shen et al., 2005b). Additionally, the cg2636 (catA) gene was cloned in E. coli and the CatA protein was purified to homogeneity. Subsequent enzyme assays demonstrated that the purified CatA protein possesses catechol 1,2-dioxygenase activity, providing evidence that both branches of the b-ketoadipate pathway operate in C. glutamicum to utilize a wide spectrum of aromatic compounds for growth. Furthermore, phenol may be converted into catechol by phenol hydroxylase activity (Shen et al., 2005b) which is most likely encoded by the cg2966 and ce2219 genes of C. glutamicum and C. efficiens, respectively (Figure 4A, B).
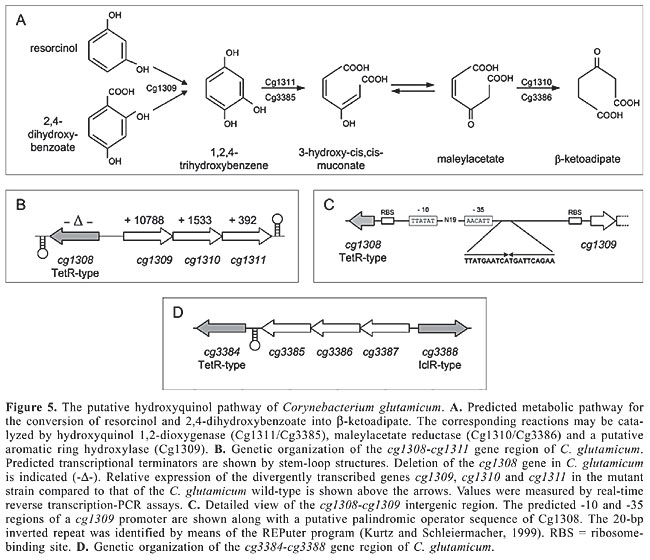
Data on the transcriptional regulation of the catechol branch of the b-ketoadipate pathway of C. glutamicum are scarce. A putative transcriptional regulator of the LuxR protein family is encoded by the cg2641 (benR) gene (Table 1; Brune et al., 2005). Since LuxR-type regulators are generally regarded to act as transcriptional activators (Bateman et al., 2002; Hansmeier et al., 2006; Cramer et al., 2006), a positive regulatory role of the BenR protein on the transcription of genes belonging to the ben and/or cat sub-clusters can be assumed. Expression of genes involved in the conversion of benzoate into catechol is also activated in Acinetobacter sp strain ADP1 and in P. putida in response to benzoate, although the regulatory proteins are members of different families and belong to the LysR and AraC/XylS regulators, respectively (Collier et al., 1998; Cowles et al., 2000). The conversion of phenol into catechol may be under positive transcriptional control since the non-syntenic coding regions cg2966 and ce2219 both are preceded by a convergently transcribed gene that encodes an AraC-type regulator (Figure 4B; Table 1). Recently, it was shown that the catA gene, encoding catechol 1,2-dioxygenase, is under direct transcriptional control by the RipA repressor, the expression of which is negatively regulated by the DtxR protein (Wennerhold et al., 2005). Transcriptional regulation by RipA is realized by the interaction of the protein with a 14-bp operator sequence that is located 337 bp upstream of the translational start codon of the catA gene but only 68 bp upstream of the benA translational start (Figure 4C). Binding of the RipA protein to this regulatory region could therefore also interfere with the expression of at least the benABCD genes (Figure 4B). However, this direct regulatory interaction of RipA with the catA-benA intergenic region apparently links catechol metabolism with the availability of iron since both proteins, RipA and DtxR, constitute a hierarchical regulatory network to control iron homeostasis in C. glutamicum at the transcriptional level (Wennerhold et al., 2005; Brune et al., 2006). The physiological link between catechol and iron metabolism is reasonable when considering that catechol 1,2-dioxygenases are iron-containing enzymes (Yamahara et al., 2002). In the presence of excess iron, the iron-sensing regulator DtxR represses transcription of the ripA gene, thereby indirectly provoking derepression of the catA gene, whereas under iron limitation, repression of ripA transcription by DtxR is relieved and catA gene expression is repressed by the synthesized RipA protein to reduce cellular iron consumption (Figure 4D). The transcriptional response of C. glutamicum to the presence of benzoate or catechol is therefore superimposed by the response of a superior regulatory network that senses the availability of iron within the cell. TRANSCRIPTIONAL REGULATION OF THE PUTATIVE HYDROXYQUINOL PATHWAYS OF CORYNEBACTERIUM GLUTAMICUM Further experiments concerning the utilization of aromatic compounds as the sole carbon and energy source showed that C. glutamicum is able to use resorcinol, 2,4-dihydroxybenzoate and 3,5-dihydroxytoluene for growth (Figure 5A; Shen et al., 2005b). Cultivation of C. glutamicum with these carbon sources was associated with hydroxyquinol 1,2-dioxygenase activity. Genome data mining revealed the presence of two gene clusters (Figure 5B, D) encoding putative hydroxyquinol 1,2-dioxygenases and maleylacetate reductases (Kalinowski et al., 2003; Shen et al., 2005b). The putative hydroxyquinol 1,2-dioxygenase genes (cg1311 and cg3385) were individually expressed in E. coli and the purified proteins showed hydroxyquinol 1,2-dioxygenase activity in enzyme assays. Whether these proteins are indeed required for the assimilation of the aforementioned aromatic compounds remains to be elucidated.
Transcriptional regulation of expression of the first gene cluster comprising cg1309 to cg1311 is apparently exerted by the TetR-type repressor Cg1308 (Table 1; Brune et al., 2005) which is encoded on the opposite DNA strand upstream of cg1309 (Figure 5B). Deletion of the cg1308 coding region in the C. glutamicum genome resulted in elevated expression of cg1309, cg1310 and cg1311, demonstrating that transcription of these genes was derepressed in the mutant strain and is thus under negative transcriptional control. A 20-bp palindromic sequence is located in the short intergenic region of cg1308 and cg1309 and may represent an appropriate operator sequence for the TetR-type repressor Cg1308 (Figure 5C). Transcriptional regulation of the second gene cluster has not been investigated so far. Two potential regulatory genes representing candidates that may be involved in controlling expression of the cg3387, cg3386 and cg3385 genes flank this gene locus (Figure 5C). The cg3384 gene encodes a repressor protein of the TetR family, whereas the cg3388 gene codes for a transcriptional regulator of the IclR family (Table 1; Brune et al., 2005). Deletion of the cg3384 coding region in the C. glutamicum genome did not affect expression of cg3385, cg3386 and cg3387, suggesting that the TetR-type repressor Cg3384 is not involved in negative regulation of the expression of the hydroxyquinol pathway genes (Brinkrolf K, Brune I and Tauch A, unpublished data). Moreover, the cg3388 gene provides the more attractive candidate for hydroxyquinol pathway regulation since genes encoding regulatory proteins of the IclR family are mostly located upstream of the target genes and are transcribed in the opposite direction (Figure 5D). The IclR-type regulator Cg3388 is somehow unusual since analysis of its protein architecture identified two winged helix DNA-binding domains and two GAF-like effector-binding domains (Gough et al., 2001; Martinez et al., 2002). Compounds acting as effectors of the Cg1308 and Cg3388 regulators await experimental identification, but could be related to resorcinol, 2,4-dihydroxybenzoate, 3,5-dihydroxytoluene, or their degradation products (Shen et al., 2005b). CONCLUSIONS AND PROSPECTS Microorganisms generally respond to changes in environmental conditions through the action of specific regulatory systems that not only detect the changes but also develop coordinated cellular responses to adapt to the new conditions. A specific transcriptional response is reasonable when, for instance, the bacterial cell is exposed to aromatic compounds that can be utilized as the sole carbon and energy source (Diaz and Prieto, 2000; Tropel and van der Meer, 2004). C. glutamicum possesses five ring-fission dioxygenases that are synthesized only upon exposure of the cells to a variety of aromatic compounds (Shen et al., 2005b). The corresponding dioxygenase genes and the entire enzymatic machinery necessary for the degradation of aromatic compounds are organized in gene clusters that are apparently under specific transcriptional control. As a result of genome data mining and microbiological studies (Brune et al., 2005; Shen et al., 2005b), we now have a profound understanding of the utilization of aromatic compounds as the sole carbon and energy source by C. glutamicum. This improvement also includes knowledge about the behavior of transcriptional regulatory proteins in the process of controlling gene expression of the respective catabolic pathways. Consequently, these data have been integrated into the CoryneRegNet database which allows the visualization of transcriptional regulatory networks by using qualitative regulatory interactions (Baumbach et al., 2006). With the exception of the catA (cg2636) gene, the regulatory interactions involved in the control of the expression of aromatic compound utilization are located separately in the transcriptional regulatory network of C. glutamicum and most likely represent single input motifs within the modular and hierarchical network structure (Shen-Orr et al., 2002; Baumbach et al., 2006). In most cases a relatively clear picture has emerged for the physiological role of the regulatory proteins that control aromatic compound degradation by C. glutamicum. The respective transcriptional regulators have been classified into the AraC, IclR, LuxR, PadR, and TetR families of regulatory proteins (Brune et al., 2005). Whereas a number of AraC, IclR and PadR family proteins are already known to be involved in transcriptional control of catabolic pathways for aromatic compounds (Gerischer, 2002; Tropel and van der Meer, 2004; Molina-Henares et al., 2006), the participation of TetR and LuxR family proteins in the regulation of aromatic compound degradation has not been widely recognized so far (Tropel and van der Meer, 2004; Ramos et al., 2005). Moreover, it was possible to identify many of the molecular components that are involved in the regulatory processes as well as the environmental signals that trigger the expression of the catabolic pathways for aromatic compounds in C. glutamicum. Both, positive and negative control mechanisms apparently enable the C. glutamicum cell to express the respective pathways only in the presence of an inducing substrate. This observation is consistent with the standard model that a positive regulator is bound to its operator site to activate gene expression, whereas the interaction of a negative regulator with its cognate DNA-binding site is relieved by derepression, resulting in elevated transcription of the target genes. However, little is currently known about the exact mechanisms of transcriptional activation and (de)repression by the identified regulatory proteins and about the interaction between these transcriptional regulators and their effector compounds. This lack of knowledge could be reduced by analyzing in vitro the DNA-binding properties of purified regulatory proteins in the presence and absence of potential effector compounds. Additionally, the elucidation of the molecular binding of experimentally identified effector compounds to purified regulatory proteins and determination of the affinity of the active regulator for its cognate operator sequence(s) may help to generate quantitative data that can be integrated into corresponding topological models of the C. glutamicum regulatory network. Accordingly, the aspired change-over from a static to a dynamic view of regulatory interactions will be an important challenge for future work in the field of genome-based systems biology of C. glutamicum. ACKNOWLEDGMENTS The authors thank Alfred Pühler and Anna-Katharina Kurze (Centrum für Biotechnologie, Universität Bielefeld) for critically reading the manuscript. REFERENCES Barthelmebs L, Lecomte B, Divies C and Cavin JF (2000). Inducible metabolism of phenolic acids in Pediococcus pentosaceus is encoded by an autoregulated operon which involves a new class of negative transcriptional regulator. J. Bacteriol. 182: 6724-6731. Bateman A, Birney E, Cerruti L, Durbin R, et al. (2002). The Pfam protein families database. Nucleic Acids Res. 30: 276-280. Baumbach J, Brinkrolf K, Czaja LF, Rahmann S, et al. (2006). CoryneRegNet: An ontology-based data warehouse of corynebacterial transcription factors and regulatory networks. BMC Genomics 7: 24. Brune I, Brinkrolf K, Kalinowski J, Puhler A, et al. (2005). The individual and common repertoire of DNA-binding transcriptional regulators of Corynebacterium glutamicum, Corynebacterium efficiens, Corynebacterium diphtheriae and Corynebacterium jeikeium deduced from the complete genome sequences. BMC Genomics 6: 86. Brune I, Werner H, Huser AT, Kalinowski J, et al. (2006). The DtxR protein acting as dual transcriptional regulator directs a global regulatory network involved in iron metabolism of Corynebacterium glutamicum. BMC Genomics 7: 21. Collier LS, Gaines GL III and Neidle EL (1998). Regulation of benzoate degradation in Acinetobacter sp. strain ADP1 by BenM, a LysR-type transcriptional activator. J. Bacteriol. 180: 2493-2501. Cowles CE, Nichols NN and Harwood CS (2000). BenR, a XylS homologue, regulates three different pathways of aromatic acid degradation in Pseudomonas putida. J. Bacteriol. 182: 6339-6346. Cramer A, Gerstmeir R, Schaffer S, Bott M, et al. (2006). Identification of RamA, a novel LuxR-type transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J. Bacteriol. 188: 2554-2567. Diaz E (2004). Bacterial degradation of aromatic pollutants: a paradigm of metabolic versatility. Int. Microbiol. 7: 173-180. Diaz E and Prieto MA (2000). Bacterial promoters triggering biodegradation of aromatic pollutants. Curr. Opin. Biotechnol. 11: 467-475. DiMarco AA and Ornston LN (1994). Regulation of p-hydroxybenzoate hydroxylase synthesis by PobR bound to an operator in Acinetobacter calcoaceticus. J. Bacteriol. 176: 4277-4284. Feng J, Che Y, Milse J, Yin YJ, et al. (2006). The gene ncgl2918 encodes a novel maleylpyruvate isomerase that needs mycothiol as cofactor and links mycothiol biosynthesis and gentisate assimilation in Corynebacterium glutamicum. J. Biol. Chem. 281: 10778-10785. Gerischer U (2002). Specific and global regulation of genes associated with the degradation of aromatic compounds in bacteria. J. Mol. Microbiol. Biotechnol. 4: 111-121. Gerischer U, Segura A and Ornston LN (1998). PcaU, a transcriptional activator of genes for protocatechuate utilization in Acinetobacter. J. Bacteriol. 180: 1512-1524. Gough J, Karplus K, Hughey R and Chothia C (2001). Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 313: 903-919. Guedon E, Jamet E and Renault P (2002). Gene regulation in Lactococcus lactis: the gap between predicted and characterized regulators. Antonie Van Leeuwenhoek 82: 93-112. Guo Z and Houghton JE (1999). PcaR-mediated activation and repression of pca genes from Pseudomonas putida are propagated by its binding to both the -35 and the -10 promoter elements. Mol. Microbiol. 32: 253-263. Gury J, Barthelmebs L, Tran NP, Divies C, et al. (2004). Cloning, deletion, and characterization of PadR, the transcriptional repressor of the phenolic acid decarboxylase-encoding padA gene of Lactobacillus plantarum. Appl. Environ. Microbiol. 70: 2146-2153. Hansmeier N, Albersmeier A, Tauch A, Damberg T, et al. (2006). The surface (S)-layer gene cspB of Corynebacterium glutamicum is transcriptionally activated by a LuxR-type regulator and located on a 6 kb genomic island absent from the type strain ATCC 13032. Microbiology 152: 923-935. Harwood CS and Parales RE (1996). The beta-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 50: 553-590. Hermann T (2003). Industrial production of amino acids by coryneform bacteria. J. Biotechnol. 104: 155-172. Herrgard MJ, Covert MW and Palsson BO (2004). Reconstruction of microbial transcriptional regulatory networks. Curr. Opin. Biotechnol. 15: 70-77. Hintner JP, Reemtsma T and Stolz A (2004). Biochemical and molecular characterization of a ring fission dioxygenase with the ability to oxidize (substituted) salicylate(s) from Pseudaminobacter salicylatoxidans. J. Biol. Chem. 279: 37250-37260. Kalinowski J, Bathe B, Bartels D, Bischoff N, et al. (2003). The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 104: 5-25. Kurtz S and Schleiermacher C (1999). REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15: 426-427. Madan Babu M and Teichmann SA (2003a). Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res. 31: 1234-1244. Madan Babu M and Teichmann SA (2003b). Functional determinants of transcription factors in Escherichia coli: protein families and binding sites. Trends Genet. 19: 75-79. Martinez SE, Beavo JA and Hol WG (2002). GAF domains: two-billion-year-old molecular switches that bind cyclic nucleotides. Mol. Interv. 2: 317-323. Martinez-Antonio A, Janga SC, Salgado H and Collado-Vides J (2006). Internal-sensing machinery directs the activity of the regulatory network in Escherichia coli. Trends Microbiol. 14: 22-27. Mason JR and Cammack R (1992). The electron-transport proteins of hydroxylating bacterial dioxygenases. Annu. Rev. Microbiol. 46: 277-305. Matic I, Taddei F and Radman M (2004). Survival versus maintenance of genetic stability: a conflict of priorities during stress. Res. Microbiol. 155: 337-341. Merkens H, Beckers G, Wirtz A and Burkovski A (2005). Vanillate metabolism in Corynebacterium glutamicum. Curr. Microbiol. 51: 59-65. Molina-Henares AJ, Krell T, Eugenia GM, Segura A, et al. (2006). Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30: 157-186. Moreno-Campuzano S, Janga SC and Perez-Rueda E (2006). Identification and analysis of DNA-binding transcription factors in Bacillus subtilis and other Firmicutes - a genomic approach. BMC Genomics 7: 147. Nishio Y, Nakamura Y, Kawarabayasi Y, Usuda Y, et al. (2003). Comparative complete genome sequence analysis of the amino acid replacements responsible for the thermostability of Corynebacterium efficiens. Genome Res. 13: 1572-1579. Patek M, Nesvera J, Guyonvarch A, Reyes O, et al. (2003). Promoters of Corynebacterium glutamicum. J. Biotechnol. 104: 311-323. Perez-Rueda E and Collado-Vides J (2000). The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res. 28: 1838-1847. Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, et al. (2005). The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69: 326-356. Resendis-Antonio O, Freyre-Gonzalez JA, Menchaca-Mendez R, Gutierrez-Rios RM, et al. (2005). Modular analysis of the transcriptional regulatory network of E. coli. Trends Genet. 21: 16-20. Romero-Steiner S, Parales RE, Harwood CS and Houghton JE (1994). Characterization of the pcaR regulatory gene from Pseudomonas putida, which is required for the complete degradation of p-hydroxybenzoate. J. Bacteriol. 176: 5771-5779. Shen X and Liu S (2005). Key enzymes of the protocatechuate branch of the beta-ketoadipate pathway for aromatic degradation in Corynebacterium glutamicum. Sci. China C Life Sci. 48: 241-249. Shen XH, Liu ZP and Liu SJ (2004). Functional identification of the gene locus (ncg12319) and characterization of catechol 1,2-dioxygenase in Corynebacterium glutamicum. Biotechnol. Lett. 26: 575-580. Shen XH, Jiang CY, Huang Y, Liu ZP, et al. (2005a). Functional identification of novel genes involved in the glutathione-independent gentisate pathway in Corynebacterium glutamicum. Appl. Environ. Microbiol. 71: 3442-3452. Shen XH, Huang Y and Liu SJ (2005b). Genomic analysis and identification of catabolic pathways for aromatic compounds in Corynebacterium glutamicum. Microbes Environ. 20: 160-167. Shen-Orr SS, Milo R, Mangan S and Alon U (2002). Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 31: 64-68. Teichmann SA and Babu MM (2004). Gene regulatory network growth by duplication. Nat. Genet. 36: 492-496. Tropel D and van der Meer JR (2004). Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol. Mol. Biol. Rev. 68: 474-500. Wennerhold J, Krug A and Bott M (2005). The AraC-type regulator RipA represses aconitase and other iron proteins from Corynebacterium under iron limitation and is itself repressed by DtxR. J. Biol. Chem. 280: 40500-40508. Xu Y, Yan DZ and Zhou NY (2006). Heterologous expression and localization of gentisate transporter Ncg12922 from Corynebacterium glutamicum ATCC 13032. Biochem. Biophys. Res. Commun. 346: 555-561. Yamahara R, Ogo S, Masuda H and Watanabe Y (2002). (Catecholato)iron(III) complexes: structural and functional models for the catechol-bound iron(III) form of catechol dioxygenases. J. Inorg. Biochem. 88: 284-294. |
|