
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
ABSTRACT. Star STING is the latest version of the STING suite of programs and corresponding database. We report on five important aspects of this package that have acquired some new characteristics, designed to add key advantages to the whole suite: 1) availability for most popular platforms and browsers, 2) introduction of the STING_DB quality assessment, 3) improvement in algorithms for calculation of three STING parameters, 4) introduction of five new STING modules, and 5) expansion of the existing modules. Star STING is freely accessible at: http://sms.cbi.cnptia.embrapa.br/SMS/, http://trantor.bioc.columbia.edu/SMS, http://www.es.embnet.org/SMS/, http://gibk26.bse.kyutech.ac.jp/SMS/ and http://www.ar.embnet.org/SMS. Key words: Protein structure analysis, Per-residue structure descriptors, Topology similarity, Structure summaries INTRODUCTION STING, a multiplatform environment for protein structure analysis, continues to build on what is considered its main asset: a principal database (DB) of per-residue-reported descriptors (available for display both numerically and graphically) for either the public protein data base (PDB) (Berman et al., 2004) or local files. Since its first appearance in 1998, STING has undergone seven major updates (Neshich et al., 2003, 2004, 2005a,b; Higa et al.,2004) with ever increasing integration of data describing the protein sequences, structure, function, and stability. We describe some new features available in the Star STING suite. PLATFORM AND BROWSER COMPATIBILITY A dual protein structure viewer capability was introduced in Star STING, with options for using either Jmol or Chime. The former makes STING compatible with major platforms and browsers. STING_DB QUALITY ASSESSMENT The STING_DB is regularly updated in synchrony with the PDB updates (once a week). All the STING_DB parameters are calculated immediately upon receiving the newly deposited structures. However, some parameters might not be successfully processed while updating STING_DB and calculating parameters in high-throughput mode. Hence, there was an immediate demand for introducing a module for checking the quality of the STING_DB. The STING_DB Quality Assessment (STING_DB QA) is an answer to this particular demand. With this new module, a user can perform tracking of the PDB files that contain insufficient information clarity for a specific structure descriptor calculation. Also, the overall quality of the STING_DB is now much easier to check, setting this STING version apart from most of the other (similar) products in this area (Galperin, 2006). ALGORITHM IMPROVEMENTS Three of the STING_DB per-residue-reported structure descriptors: Order of Cross-Link, Order of Cross-Presence as well as Evolutionary Pressure, were re-calculated, using new default input parameter settings for the corresponding programs. The cross-links were re-defined in STING; we previously defined them as the contacts (any type from possible five classes) established among residues that are far apart in the protein primary sequence, but are close in its 3-D folding. Only a single occurrence is counted for the Order of Cross-Link, even though several contacts could be identified, starting from the central amino acid, which can make more than one contact, and each of these can be established with a different amino acid belonging to the same stretch of probing sequence size [15, 20 and 30 residues long]. We introduced an additional restriction in Star STING, so that the occurrence of the contact is only counted for the order of cross-link if the target residues are also 15, 20 or 30 amino acids (respectively) apart in a primary sequence. As a result, we have fewer occurrences of identified cross-links. This further emphasizes the importance of those that remain. We have also used a new Rate4Site algorithm for calculating the Evolutionary Pressure parameter. The new version of this algorithm was used with the following default input values: no branch length optimization for phylogenetic trees and 10 discrete “Gamma categories” (Mayrose et al., 2004). This algorithm configuration allowed us to obtain the best balance between accuracy of reported data and running time on available CPUs, which in turn will allow us to make more frequent STING_DB updates. NEW MODULES FOR COMPLEX PROTEIN STRUCTURE ANALYSIS Five new STING modules were introduced: I. Multiple Structures - Single Parameter 2-D plot for comparing a chosen parameter among several structures (Figure 1, inset H); II. STING_TGZ for obtaining encapsulated STING_DB parameters for up to five public PDB files at the same time; III. Contact Distance Map for simultaneous visualization of the five classes of contacts for all amino acids of a chosen structure; IV. TopSiMap for evaluating the topology similarity among any two structures, based on the contact pattern (Figure 1, inset E), and V. Protein Contacts Difference, which generates an easy-to-grasp HTML table listing contact differences among any two structures (Figure 1, inset F). 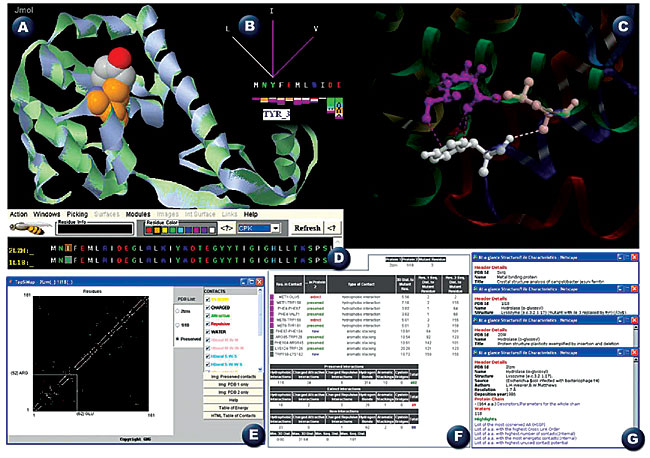 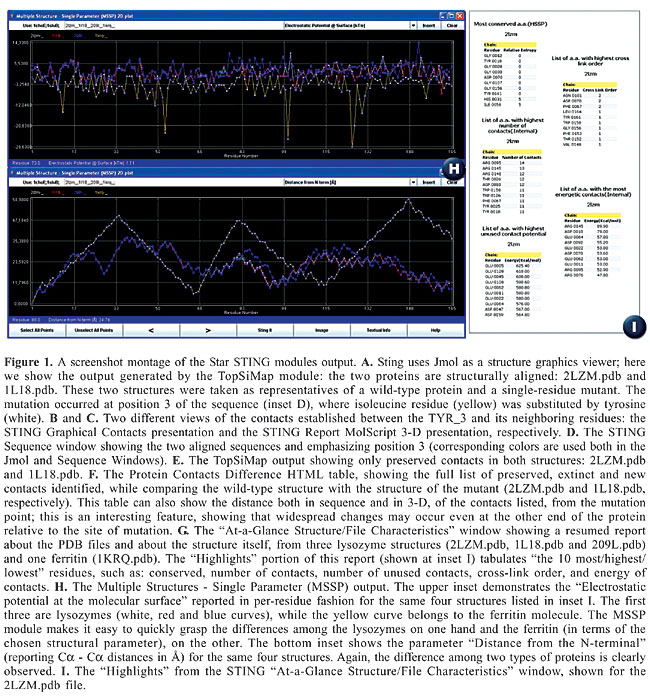 GENERAL IMPROVEMENTS In Star STING we improved: 1) The procedure for selecting the structure parameter ranges in JavaProtein Dossier while searching for those amino acids that satisfy particular conditions. Most importantly, the parameter ranges could now be saved and transferred from one STING session to the other, greatly facilitating the process of selection. 2) In addition, the STING Report module was expanded with new module images, including MolScript (Kraulis, 1991) presentation of the amino acid whose characteristics are particularly targeted (Figure 1, inset C). 3) At the opening of STING, a user is now presented with a third window: “At-a-glance Structure/File Characteristics” (Figure 1, inset G), offering general information on this specific PDB entry, facilitating analysis by displaying some background information, summaries about the structure and the “Highlights” - the list of residues with some structural descriptors occupying an outstanding position among the others (Figure 1, inset I). FUTURE DEVELOPMENTS The Star STING and STING_DB are already being expanded: two new parameters will be added in Blue Star STING (protein-ligand contacts and co-evolving amino acids). The Blue Star STING will have the following new modules: 1) 3-D Java Protein Interface Viewer, 2) 2-D Interface Maps, 3) Multiple Parameter 3-D Plot, 4) Sting Enzyme Classification, 5) Protein Ligand Contacts, 6) Topologs 100, 7) Topologs Astral 40, and 8) AA Co-evolution. All these new modules will further advance the capabilities of the STING environment toward ever more complex studies and analyses of protein structure. ACKNOWLEDGMENTS The authors express special thanks to Jose Valverde from CNB, Madrid, Spain; Oscar Grau from LaPlata University in Argentina; Megan Restuccia from Columbia University in New York City, and Akinori Sarai from Kyushu Technical University, for their collaboration in testing and maintaining STING at the respective STING mirror sites. We also thank Nir Ben-Tal and his group for aiding us in implementing Rate4Site software and indicating the best input parameters for calculating the evolutionary pressure parameter. Research supported by CNPq/Brazil, under grant No. 401695/2003-4. REFERENCES Berman HM, Bourne PE and Westbrook J (2004). The PDB: a case study in management of community data. Curr. Proteomics 1: 49-57. Galperin MY (2006). The molecular biology database collection: 2006 update. Nucleic Acids Res. 34: D3-D5. Higa RH, Togawa RC, Montagner AJ, Palandrani JC, et al. (2004). STING Millennium Suite: integrated software for extensive analyses of 3-D structures of proteins and their complexes. BMC Bioinformatics 5: 107. Kraulis PJ (1991). MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24: 946-950. Mayrose I, Mitchell A and Pupko T (2004). Site-specific evolutionary rate inference: taking phylogenetic uncertainty into account. J. Mol. Evol. 60: 345-353. Neshich G, Togawa RC, Mancini AL, Kuser PR, et al. (2003). STING Millennium: a web-based suite of programs for comprehensive and simultaneous analysis of protein structure and sequence. Nucleic Acids Res. 31: 3386-3392. Neshich G, Rocchia W, Mancini AL, Yamagishi ME, et al. (2004). JavaProtein Dossier: a novel web-based data visualization tool for comprehensive analysis of protein structure. Nucleic Acids Res. 32: W595-W601. Neshich G, Mancini AL, Yamagishi ME, Kuser PR, et al. (2005a). STING Report: convenient web-based application for graphic and tabular presentations of protein sequence, structure and function descriptors from the STING database. Nucleic Acids Res. 33: D269-D274. Neshich G, Borro LC, Higa RH, Kuser PR, et al. (2005b). The Diamond STING server. Nucleic Acids Res. 33: W29-W35. |
|