![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
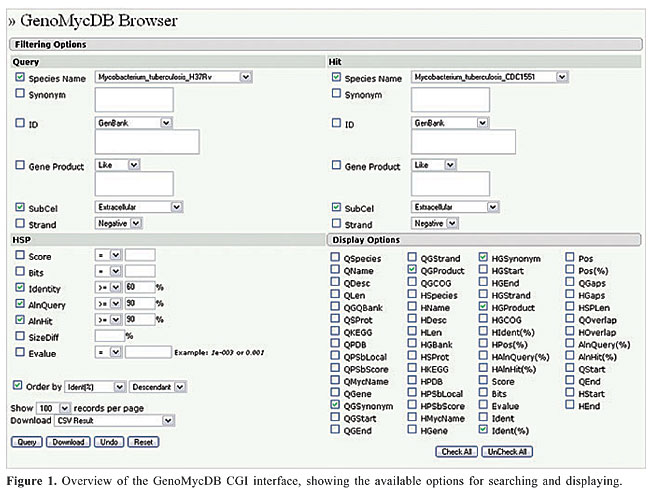
ABSTRACT. Several databases and computational tools have been created with the aim of organizing, integrating and analyzing the wealth of information generated by large-scale sequencing projects of mycobacterial genomes and those of other organisms. However, with very few exceptions, these databases and tools do not allow for massive and/or dynamic comparison of these data. GenoMycDB (http://www.dbbm.fiocruz.br/GenoMycDB) is a relational database built for large-scale comparative analyses of completely sequenced mycobacterial genomes, based on their predicted protein content. Its central structure is composed of the results obtained after pair-wise sequence alignments among all the predicted proteins coded by the genomes of six mycobacteria: Mycobacterium tuberculosis (strains H37Rv and CDC1551), M. bovis AF2122/97, M. avium subsp. paratuberculosis K10, M. leprae TN, and M. smegmatis MC2 155. The database stores the computed similarity parameters of every aligned pair, providing for each protein sequence the predicted subcellular localization, the assigned cluster of orthologous groups, the features of the corresponding gene, and links to several important databases. Tables containing pairs or groups of potential homologs between selected species/strains can be produced dynamically by user-defined criteria, based on one or multiple sequence similarity parameters. In addition, searches can be restricted according to the predicted subcellular localization of the protein, the DNA strand of the corresponding gene and/or the description of the protein. Massive data search and/or retrieval are available, and different ways of exporting the result are offered. GenoMycDB provides an on-line resource for the functional classification of mycobacterial proteins as well as for the analysis of genome structure, organization, and evolution. Key words: Mycobacteria, Genome evolution, Perl programming, Functional classification, FASTA, MySQL INTRODUCTION Complete genome sequences are a unique source of data, because together with the epigenetic networks and through their interaction with such networks they represent in principle all the necessary information to make an organism. However, it is not immediately obvious what we can do with all this information. For instance, it is believed that the comprehensive analysis of entire genomes has the potential to provide a complete understanding of the genetics, biochemistry, physiology, and pathogenesis of microorganisms (Brosch et al., 2001). In contrast, it is argued that such potential can only be realized by the comparative study of genomes, syntenic regions or genes of two or more species, subspecies or strains, because a genome considered alone, without the phylogenetic framework of the evolutionary process, merely provides an incomplete understanding of those issues (Clark, 1999). In the case of pathogenic microorganisms, especially mycobacteria, numerous potential applications of comparative genome analysis have been reported, aimed particularly at the prevention, treatment, and diagnosis of tuberculosis and other mycobacterial diseases, including i) metabolic reconstruction and identification of unique genes and virulence factors (Gordon et al., 2002), ii) characterization of pathogens and identification of new diagnostic and therapeutic targets (Fitzgerald and Musser, 2001), iii) investigation of the molecular basis of differences in pathogenesis, host range and phenotypes between clinical isolates and natural populations of pathogens (Behr et al., 1999; Brosch et al., 2001; Kato-Maeda et al., 2001; Cole, 2002), and iv) investigation of the genetic basis of virulence and drug resistance in tuberculosis-causing bacteria (Randhawa and Bishai, 2002). With the aim of providing an on-line resource for the functional classification of mycobacterial proteins as well as for the analysis of the genome structure, organization and evolution in such species, we developed GenoMycDB, a relational database for large-scale comparative analyses of completely sequenced mycobacterial genomes based on their predicted protein content. This system presents many important advantages over similar databases, such as flexibility, scalability and cross-referencing. MATERIAL AND METHODS Currently, GenoMycDB comprises the result obtained with pair-wise sequence alignments among all predicted proteins coded by the genomes of five pathogenic mycobacteria and one opportunist, respectively: Mycobacterium tuberculosis (strains H37Rv and CDC1551) - the causative agent of human tuberculosis; M. bovis (strain AF2122/97) - the etiological agent of tuberculosis in cattle and many other mammals, including humans; M. avium subsp. paratuberculosis (strain K10) - the etiological agent of paratuberculosis in ruminant animals, also implicated as the etiological agent of Crohn’s disease in humans; M. leprae (strain TN) - the causative agent of leprosy, and M. smegmatis (strain MC2 155) - a saprophyte, usually non-pathogenic. The database stores the computed similarity parameters of every aligned pair, providing for each protein sequence the predicted subcellular localization, the assigned COG(s) (cluster of orthologous groups), the description of the corresponding gene, and links to several important databases: GenBank (Benson et al., 2005), SwissProt/TrEMBL (Boeckmann et al., 2003), PDB (Berman et al., 2000), KEGG (Kanehisa, 1997; Kanehisa and Goto, 2000), and 2D-PAGE at the Max Planck Institute for Infection Biology (Pleissner et al., 2004). GenoMycDB was implemented in MySQL, version 4.0.24 (http://www.mysql.com/), a high-performance but relatively simple database management system, freely available for most in-house uses (Dubois, 2000), and its graphical CGI interface, GenoMycDB Browser, was programmed in Perl, version 5.8.4 (http://www.perl.org/; Figure 1).
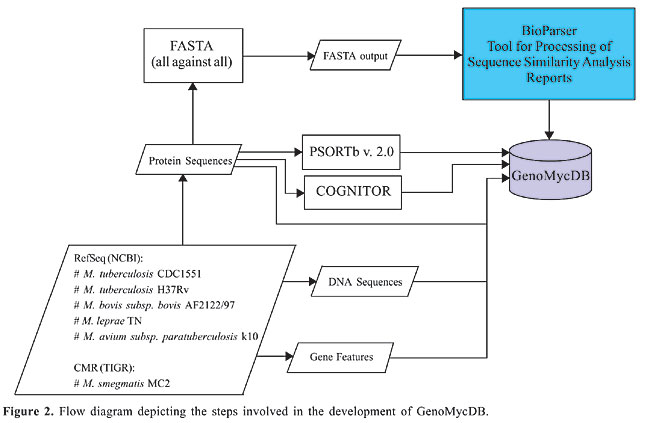
The predicted protein sequences coded by the genomes of the aforementioned mycobacteria and the features of their corresponding genes were obtained from the Reference Sequence (RefSeq) database (http://www.ncbi.nlm.nih.gov/RefSeq/) (Pruitt et al., 2000, 2005; Pruitt and Maglott, 2001) at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/) and, exclusively for M. smegmatis MC2 155, from the Comprehensive Microbial Resource database (http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi) (Peterson et al., 2001) at the Institute for Genomic Research (http://www.tigr.org/). The compiled protein data set (24,835 sequences) was submitted to three different analyses, providing most of the GenoMycDB data source (Figure 2): i) an all against all sequence comparison using the FASTA similarity search program (Pearson and Lipman, 1988; Pearson, 1990) version 3.4t21 (ftp://ftp.virginia.edu/pub/fasta/), with the program default parameters (ktup = 2, optimized score = 16, gap opening penalty = -10, gap extension penalty = -2, matrix = BLOSUM50, filter = 0, e-value cutoff = 10); ii) the computational prediction of the subcellular localization of the proteins using the PSORTb program (Gardy et al., 2003, 2005), version 2.0.2 (http://www.psort.org/downloads/index.html), employing the model built for Gram-positive bacteria, and iii) the assignment of the proteins to COG(s) using the COGNITOR program (Tatusov et al., 2000) (xugnitor.c - ftp://ftp.ncbi.nih.gov/pub/COG/old/util/), making use of a previously described method for the classification of new sequences in pre-existing COG(s) (Tatusov et al., 1997, 2000).
FASTA was chosen to perform the sequence comparison because it is faster than implementations of the Smith-Waterman algorithm (Smith and Waterman, 1981), thus guaranteeing the finding of a mathematically optimal (highest scoring) solution, exhibiting almost the same sensitivity by default. The number of alignments achieved with such a comparison was exactly 1,452,022, excluding self-comparisons. The results of the PSORTb and COGNITOR analyses are summarized in Table 1. Overall, 13,514 proteins of our dataset were assigned to pre-existing COGs. For each genome, approximately 64-74% of the predicted proteome could be assigned to COGs, except for M. smegmatis, for which only 13.1% of the total predicted proteins could be attributed to COGs. Since the genome annotation of this opportunist is still in progress (http://www.tigr.org/tdb/mdb/mdbinprogress.html), it is possible that the low fraction of proteins assigned to COGs is due to open reading frame prediction errors (such as frame shift) in the annotation process. The subcellular localization prediction also showed variations among these species. The most significant variations occurred in the fraction of proteins predicted to be extracellular. M. avium and M. leprae exhibited the lowest fractions (1.56 and 1.93%, respectively), followed by M. smegmatis (2.5%), and by M. tuberculosis H37Rv and M. bovis (approximately 3.5% for both). The M. tuberculosis CDC1551 strain gave the highest fraction of predicted extracellular proteins (5.06%), approximately 1.5% more than in the M. tuberculosis H37Rv strain genome.
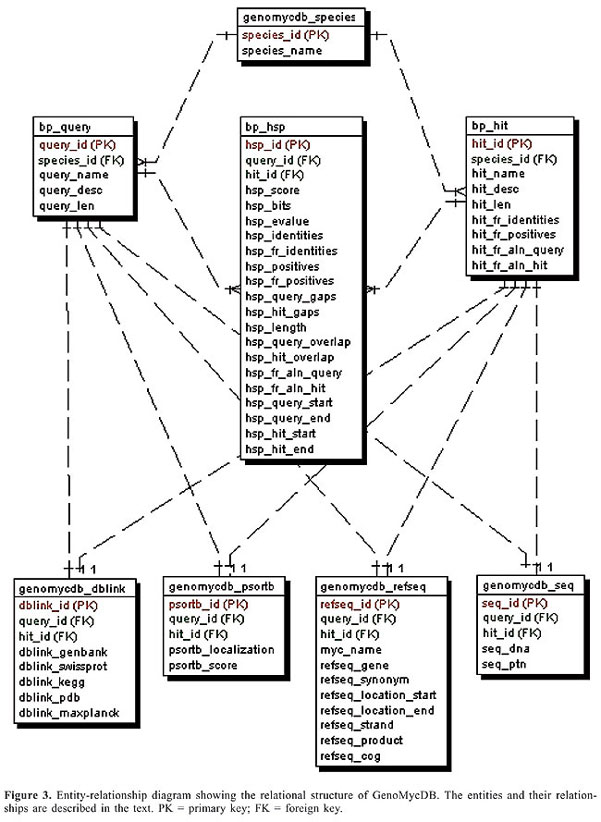
The FASTA output file was analyzed with the BioParser program (Catanho et al., 2006) (http://www.dbbm.fiocruz.br/BioParser.html), a tool designed for the processing of sequence similarity analysis reports; the results were parsed and automatically stored in a local MySQL database, comprising the central structure of the GenoMycDB: tables bp_query, bp_hit and bp_hsp (Figures 2 and 3).
The proposed structure is simple and intuitive; for each aligned pair present in the sequence similarity report, the attributes related to the query and hit sequences are stored (without redundancy) in the bp_query and bp_hit tables, respectively. The attributes that characterize each alignment, otherwise known as HSP (high scoring pair), are stored in the bp_hsp table, which is linked to the query and hit tables by two foreign keys: query_id and hit_id, respectively (Catanho et al., 2006). Five additional tables were included in GenoMycDB (Figures 2 and 3), containing the following data/information: · genomycdb_species - comprises the scientific name of each species/strain represented in GenoMycDB (species_name); · genomycdb_dblink - includes the identifying numbers of each mycobacterial protein sequence in the following databases: GenBank (dblink_genbank), SwissProt/TrEMBL (dblink_swissprot), PDB (dblink_pdb), KEGG (dblink_kegg), and 2D-PAGE at the Max Planck Institute for Infection Biology (dblink_maxplanck); · genomycdb_psortb - consists of the predicted subcellular localization of each protein (psortb_localization) and the score obtained in the prediction analysis (psortb_score); · genomycdb_refseq - provides for each mycobacterial protein: the GenoMycDB derivative name (the species name followed by a sequential number representing the relative position in the genome of the corresponding gene from the origin of replication) (myc_name), the name of the corresponding gene (refseq_gene), the synonym of the gene (refseq_synonym), the localization of the gene in the genome (refseq_location_start, refseq_location_end, and refseq_strand), the protein description (refseq_product), and the assigned COG(s) (refseq_cog); · genomycdb_seq - provides the protein (seq_ptn), and DNA (seq_dna) sequence of each mycobacterial protein. All these five tables are linked to the bp_query and bp_hit tables by the query_id and hit_id foreign keys, respectively. The bp_query and bp_hit tables are linked to the genomycdb_species table by the species_id foreign key (Figure 3). RESULTS GenoMycDB was designed for large-scale comparative analysis, offering a variety of searching/retrieving methods (Figures 1 and 3). The selection of aligned pairs with specific attributes can be done i) based on one or multiple alignment parameters (section Filtering Options, sub-section HSP) - raw score (Score); bit score (Bits); fraction of identical positions for a given HSP (Identity%); fraction of the query and/or hit sequence that has been aligned within a given HSP (AlnQuery% and AlnHit%, respectively); difference in length, expressed as a fraction, between the query and hit sequences (SizeDiff), and number of alignments expected by chance (Evalue) - and/or ii) based on one or multiple features characterizing one or both sequences of the aligned pair (section Filtering Options, sub-sections Query and Hit) - species name (Species Name); synonym of the corresponding gene(s) (Synonym); identifying number(s) of the protein(s) in the GenBank, KEGG, PDB or SwissProt/TrEMBL database (Id); presence or absence of a given key word in the protein description (Gene Product); predicted subcellular localization of the protein (SubCel), and DNA strand where the corresponding gene is located (Strand). Users can conveniently choose one field or a combination of fields to formulate the search, taking into account that a logical AND connects all these fields to each other. The Display Options section exhibits all available attributes that can be selected to compose the result (Table 2).       The result of each search is displayed as a table, in which each line corresponds to a particular alignment, and each column represents a sequence or an alignment attribute (Figure 4). The first columns, namely, Tools, Fasta, QLinks, and HLinks, offer different means to analyze a selected sequence or pair of sequences individually; it is possible to execute a global alignment between the sequences using the CLUSTAL W program (Thompson et al., 1994) (http://www.ebi.ac.uk/clustalw/), at both levels: protein and DNA (ClustalW); in addition, one can visualize the sequence(s) in the FASTA format (QSeq and HSeq), or access the page(s) of the sequence(s) in other database(s) (GBank, SProt, KEGG, PDB, or MPlanck). There are two different ways to export the result: i) save the selected records displayed in the browser or all records returned in a table format flat file, choosing the CVS Result option in the Download drop-down button of the page containing the result (Figure 4) or in the similar button of the GenoMycDB Browser main page (Figure 1), respectively, and ii) save the sequences (DNA or protein) of the selected pairs or the whole sequence set (DNA or protein) corresponding to all records returned in a FASTA format flat file, choosing the appropriate option (Query DNA Sequences, Query Protein Sequences, Hit DNA Sequences, or Hit Protein Sequences) in the same Download drop-down buttons and pages.
In summary, GenoMycDB provides an on-line resource for large-scale comparative analysis of completely sequenced mycobacterial genomes based on their predicted protein content. Through the GenoMycDB Browser, users can dynamically select pairs or groups of potential homologs between selected species/strains based on different aspects of similarity between the aligned sequences and/or on particular features characterizing one or both sequences of the aligned pair. One or multiple alignment parameters can be defined to establish a reliable cutoff of similarity to infer homology. Links to several important databases are dynamically produced for each record in the customized searching result, expanding and facilitating the analysis of the data. Sequences (both protein and DNA) of individually selected records can be globally aligned, allowing more detailed examination of the compared pair. Different ways of exporting and visualizing the results are offered, making it easier to process and analyze the information. DISCUSSION The application of comparative genomic methods for the study of pathogenic microorganisms has been successfully explored, especially in mycobacteria. Several databases and computational tools have been created, aiming to organize, integrate and analyze the wealth of information generated by large-scale sequencing projects of mycobacterial genomes and other organisms (http://genolist.pasteur.fr/; http://myco.bham.ac.uk/). However, with very few exceptions (Uchiyama, 2003; Choi et al., 2005), these databases and tools do not allow massive and/or dynamic comparisons of such data. Usually, searches in these databases are genome-guided, and comparisons between genomes/genes are either pre-computed or manually accomplished, since the provided datasets are not related to each other. In addition, the parameters employed to compare the data are commonly pre-defined, giving little or no freedom to the user. Some of them have outputs that are quite difficult to interpret, and inconsistent sequence annotation is another relevant problem. As demonstrated in Results, GenoMycDB overcomes the aforementioned problems, offering a flexible, scalable, functional, cross-referenced, and user-friendly system for the comparative genomic analyses of representatives of the genus Mycobacterium. Furthermore, the same structure and database interface can easily be applied to other groups of genomes, extending the potential of our system. In our laboratory, GenoMycDB is currently being used to study the nucleotide evolutionary rates among protein-coding regions of mycobacteria, to analyze point mutations and polymorphisms among selected protein-coding regions of M. tuberculosis complex species, and to investigate the factors shaping codon usage in mycobacteria. In addition, the database is presently being used to annotate the genome of BCG Moreau, a vaccine strain derived from M. bovis used to prevent tuberculosis in the Brazilian population; this bacterium is being sequenced in our laboratory (gap closure phase). Therefore, GenoMycDB provides a valuable tool for the comparative analyses of mycobacterial genomes, making it possible to identify evolutionary, structural, and functional relationships between proteins in such genomes. Future developments include new search fields, logical operators, sequence analysis and visualization tools, new sequenced mycobacterial genomes, and additional sequence features. ACKNOWLEDGMENTS We thank CNPq, PAPES-FIOCRUZ, WHO/TDR, UNU-BIOLAC LacBioNet, and CYTED-RIB for support. REFERENCES Behr MA, Wilson MA, Gill WP, Salamon H, et al. (1999). Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284: 1520-1523. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. (2005). GenBank. Nucleic Acids Res. 33: D34-D38. Berman HM, Westbrook J, Feng Z, Gilliland G, et al. (2000). The Protein Data Bank. Nucleic Acids Res. 28: 235-242. Boeckmann B, Bairoch A, Apweiler R, Blatter MC, et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31: 365-370. Brosch R, Pym AS, Gordon SV and Cole ST (2001). The evolution of mycobacterial pathogenicity: clues from comparative genomics. Trends Microbiol. 9: 452-458. Catanho M, Mascarenhas D, Degrave W and de Miranda AB (2006). BioParser: A tool for processing of sequence similarity analysis reports. Appl. Bioinformatics (in press). Choi K, Ma Y, Choi JH and Kim S (2005). PLATCOM: a Platform for Computational Comparative Genomics. Bioinformatics 21: 2514-2516. Clark MS (1999). Comparative genomics: the key to understanding the Human Genome Project. Bioessays 21: 121-130. Cole ST (2002). Comparative and functional genomics of the Mycobacterium tuberculosis complex. Microbiology 148: 2919-2928. Dubois P (2000). MySQL. New Riders Publishing, Indianapolis, IN, USA. Fitzgerald JR and Musser JM (2001). Evolutionary genomics of pathogenic bacteria. Trends Microbiol. 9: 547-553. Gardy JL, Spencer C, Wang K, Ester M, et al. (2003). PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 31: 3613-3617. Gardy JL, Laird MR, Chen F, Rey S, et al. (2005). PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 21: 617-623. Gordon SV, Brosch R, Eiglmeier K, Garnier T, et al. (2002). Royal Society of Tropical Medicine and Hygiene Meeting at Manson House, London, 18th January 2001. Pathogen genomes and human health. Mycobacterial genomics. Trans. R. Soc. Trop. Med. Hyg. 96: 1-6. Kanehisa M (1997). A database for post-genome analysis. Trends Genet. 13: 375-376. Kanehisa M and Goto S (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28: 27-30. Kato-Maeda M, Rhee JT, Gingeras TR, Salamon H, et al. (2001). Comparing genomes within the species Mycobacterium tuberculosis. Genome Res. 11: 547-554. Pearson WR (1990). Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 183: 63-98. Pearson WR and Lipman DJ (1988). Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85: 2444-2448. Peterson JD, Umayam LA, Dickinson T, Hickey EK, et al. (2001). The comprehensive microbial resource. Nucleic Acids Res. 29: 123-125. Pleissner KP, Eifert T, Buettner S, Schmidt F, et al. (2004). Web-accessible proteome databases for microbial research. Proteomics 4: 1305-1313. Pruitt KD and Maglott DR (2001). RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29: 137-140. Pruitt KD, Katz KS, Sicotte H and Maglott DR (2000). Introducing RefSeq and LocusLink: curated human genome resources at the NCBI. Trends Genet. 16: 44-47. Pruitt KD, Tatusova T and Maglott DR (2005). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33: D501-D504. Randhawa GS and Bishai WR (2002). Beneficial impact of genome projects on tuberculosis control. Infect. Dis. Clin. North Am. 16: 145-161. Smith TF and Waterman MS (1981). Comparison of biosequences. Adv. Appl. Math. 2: 482-489. Tatusov RL, Koonin EV and Lipman DJ (1997). A genomic perspective on protein families. Science 278: 631-637. Tatusov RL, Galperin MY, Natale DA and Koonin EV (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28: 33-36. Thompson JD, Higgins DG and Gibson TJ (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673-4680. Uchiyama I (2003). MBGD: microbial genome database for comparative analysis. Nucleic Acids Res. 31: 58-62. |
|