![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
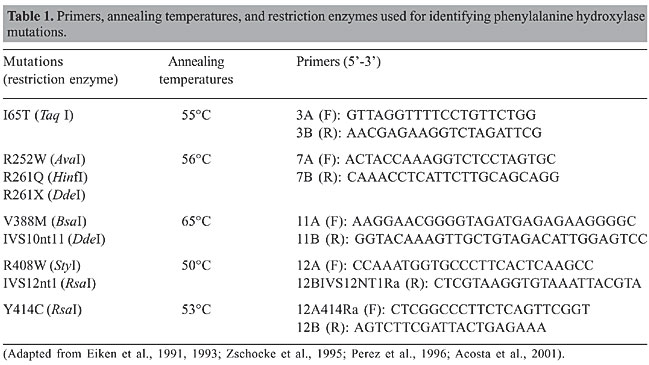
ABSTRACT. In order to determine the phenylketonuria (PKU) mutation spectrum in the population of Minas Gerais State, Brazil, 78 unrelated PKU patients found by the neonatal screening program from 1993 to 2003 were tested for nine phenylalanine hydroxylase mutations. These mutations were selected due to their high frequencies in other Brazilian populations and in Portugal, where the largest contingent of the Caucasian component of the Brazilian population originated from. The most frequent mutations were V388M (21%), R261Q (16%), IVS10nt11 (13.4%), I65T (5.7%), and R252W (5%). The frequencies of the other four mutations (R261X, R408W, Y414C, and IVS12nt1) did not reach 2%. By testing these nine mutations, we were able to identify 64% of the PKU alleles in our sample. V388M frequency was higher than in any other known population and almost three times larger than that observed in Portugal, probably reflecting genetic drift. The mutation profile, as well as the relative frequency of the different mutations, suggest that the Minas Gerais population more closely resembles that of Portugal than do the other Brazilian populations that have already been tested. Key words: Phenylalanine hydroxylase, Phenylketonuria, Mutation screening INTRODUCTION Phenylketonuria (PKU; OMIM #261600) is an autosomal recessive disorder caused by a deficiency of phenylalanine hydroxylase (PAH, EC 1.14.16.1). PAH catalyses the hydroxylation of phenylalanine to tyrosine. If it is precociously introduced, a low-phenylalanine diet prevents the mental retardation associated with PKU (MacDonald, 2000; Fisch, 2000; Scriver and Kaufman, 2001). Consequently, neonatal screening programs have constantly targeted PKU (McCabe et al., 2002; Carreiro-Lewandowski, 2002). Currently, more than 490 PAH mutations have been described (PAH Mutation Analysis Consortium; http://www.pahdb.mcgill.ca, Scriver et al., 2003). PAH mutation frequencies show strong variation among populations. These differences are attributable to the history of the populations (Hofman et al., 1991; Perez et al., 1993, 1999; Guldberg et al., 1998; Yang et al., 2001; Zschocke, 2003). Due to the large number of mutations, most of the patients are compound heterozygotes, which explains at least partially the considerable phenotypic variability observed in this disease. Determination of the PAH mutations in each patient can help predict the severity of the disease (Rivera et al., 1998; Benit et al., 1999; Pey and Martinez, 2005). Depending on the mutation, some patients are more efficiently treated with BH4, the PAH co-factor (Bernegger and Blau, 2002; Blau and Erlandsen, 2004; Steinfeld et al., 2004; Cerone et al., 2004). Minas Gerais is a state with 16 million inhabitants in southeastern Brazil. A neonatal screening program has been conducted in Minas Gerais since 1993 by the Núcleo de Pesquisa em Apoio ao Diagnóstico (NUPAD) of the Universidade Federal de Minas Gerais. Monthly, approximately 20,000 newborns are tested for PKU, hypothyroidism and sickle cell disease. The mean coverage is approximately 94% of the births in this state. PKU incidence in Minas Gerais has been estimated to be about 1:20,000 (Serjeant, 2000). This frequency is somewhat lower than previously reported figures for Caucasians (1:10,000) and Orientals (1:16,500; Scriver and Kaufman, 2001). We sought to develop a cost-efficient strategy for PAH mutation screening in Minas Gerais. We selected for evaluation those mutations that were highly prevalent in Western Europe, particularly in Portugal, historically known to be the main source of the Caucasian component of the Brazilian population. Also, mutations found to be frequent in two other Brazilian subpopulations, those from São Paulo (Acosta et al., 2001) and from the two southernmost Brazilian states, Santa Catarina and Rio Grande do Sul (Santana da Silva et al., 2003), were added to the project. Initially, we selected a set of nine mutations. PATIENTS AND METHODS We examined 78 unrelated patients sequentially diagnosed by a neonatal screening program. Prior to the beginning of the investigation, the project was approved by the Ethics Committee on Research Board of the Universidade Federal de Minas Gerais. Patients were screened for the presence of the PAH mutations I65T, R252W, R261Q, R261X, IVS10nt11, V388M, R408W, Y414C, and IVS12nt1. A blood sample (5 mL) was collected from each patient, after obtaining informed consent from his or her parents or from a legally designated guardian. Genomic DNA was isolated, as previously described (Miller et al., 1988). PCR amplification was performed for the fragments of the PAH gene in which these mutations are located. Primers, annealing temperatures, and restriction enzymes used for digesting the PCR products are shown in Table 1. Restriction fragments were separated by polyacrylamide gel electrophoresis (Figure 1). Gels were stained with silver nitrate or with ethidium bromide.
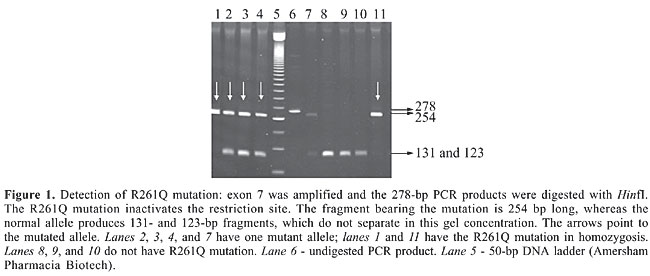
RESULTS The most common mutations among the newborns were V388M (21%), R261Q (16%), IVS10nt11 (13.4%), I65T (5.7%), and R252W (5%). Individually, the other four mutations (R261X, R408W, Y414C, and IVS12nt1) had frequencies lower than 2%. By screening these nine mutations we were able to identify 100/156 alleles, corresponding to 64% of the PKU alleles in this sample (Table 2).
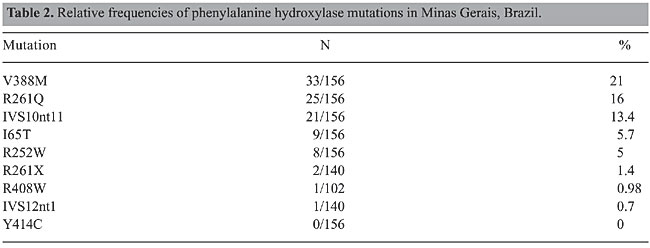
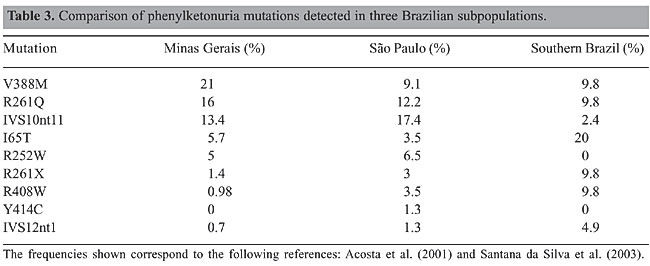
Among these 100 alleles, 38 were found in homozygosis and 62 in heterozygosis. Complete PAH locus genotyping was established for 39 of 78 PKU patients (50%). A further 22 PKU patients (28%) had one of the PKU alleles identified. Consanguinity between the patient’s parents was reported in 16/72 (22%) of the unrelated patients for whom information was available. Consanguineous marriages were reported by 54% of the parents of the true homozygous vs 3% of the parents of the compound heterozygous patients. This difference is significant (c2 = 13.84; P < 0.005). Despite the high consanguinity values, we could not determine PAH allele frequencies for this sample, as we did not have an estimation of F (Sokal and Rohlf, 1969). Maps of the geographical distribution of the PKU patients and of PAH alleles in the State of Minas Gerais were created with Tabwin programs (developed by SUS - Sistema Único de Saúde, Brazil, freely available at http://www.datasus.gov.br). There was no apparent geographic aggregation within the state, both for PKU patients and for the mutations (data not shown). DISCUSSION Three main groups have contributed to the Brazilian population: Amerindian, African, and Caucasian, the latter being predominantly Portuguese. However, there is a considerable variation in the contribution of each group to the ethnic admixture in the different regions of the country. Moreover, the three components are heterogeneous. At the time of the arrival of Europeans in Brazil, in 1500 A.D., Tupi-Guarani Amerindians occupied the region that nowadays constitutes the State of Minas Gerais. Although there were occasional incursions, the first Portuguese settlements in the region were established only after 1693, when gold mines were discovered. During the next 150 years, succeeding economic cycles based on diamonds, milk, coffee, and iron brought African slaves to Minas Gerais, mainly from Guinea-Bissau and Angola. The influx of African slaves diminished during the first fifty years of the 19th century and was followed by migration from Europe, North Africa, and East Asia. When compared to Minas Gerais, São Paulo, and particularly the southern Brazilian states, received a proportionately smaller African and a larger and more diversified European contingent (Bortolini et al., 1997; Alves-Silva et al., 2000; Carvalho-Silva et al., 2001; Bortolini et al., 2003; Callegari-Jacques et al., 2003). The lower PKU incidence ascertained by the neonatal screening program in Minas Gerais, approximately 1:20,000, probably reflects the higher contribution of African ancestry to this population. Differences in peopling among Brazilian regions after 1500 A.D. are strong enough to bring variations in allele frequencies, and consequently, each Brazilian state must establish its own strategy for a mutation-screening program. In order to begin PKU mutation screening in Minas Gerais State we chose a set of nine mutations that can be easily screened by PCR amplification, which are prevalent in São Paulo, in the southern Brazilian states or in Portugal, followed by a restriction digest. We compared the frequencies of the mutations detected in Minas Gerais to those observed in São Paulo and southern Brazil (Table 3).
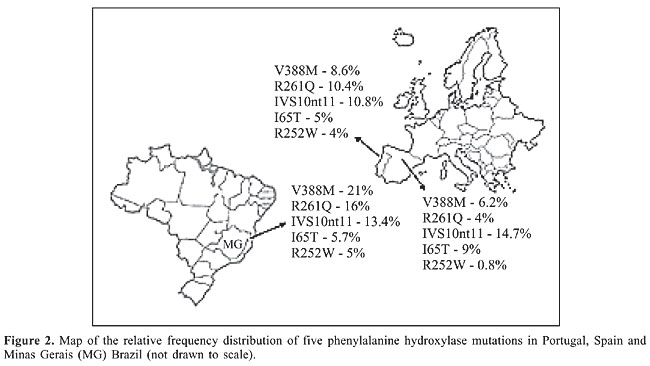
Phenylketonuria mutations in Minas Gerais vs Portugal and other European populations The frequency found for V388M in Minas Gerais is the highest described so far in the world. This mutation accounts for 8.6% of the PKU alleles in Portugal and 6.2% in Spain (Figure 2). V388M is virtually absent in other European populations, and consequently it has been referred to as a Spanish or Iberian mutation (Desviat et al., 1995; Perez et al., 1996, 1997; Rivera et al., 1998). Coincidentally, the frequency of V388M in Chile (13%) is also higher than that observed in Spain (Perez et al., 1999). There is a large variation in the frequency of the R261Q mutation in Caucasian populations, varying from 1.2% in Ireland to 32% in Switzerland, its probable place of origin, with a mean European frequency of 4%. The frequency of this mutation was somewhat higher in Minas Gerais than in Portugal (10.4%; Eisensmith and Woo, 1992; Zschocke et al., 1995; Rivera et al., 1998; Desviat et al., 1999; Zschocke, 2003). The other mutation, IVS10nt11, is also distributed throughout Europe with frequencies varying from 1 to 25% (Zschocke, 2003). Its frequency in Minas Gerais (13.4%) is similar to those described in Portugal (10.8%) and Spain (14.7%; Perez et al., 1997; Rivera et al., 1998). The I65T mutation, one of the three most common mutations in the Iberian Peninsula, also has a similar frequency in Minas Gerais (5.7%) and in other Latin American populations, as with the other PKU mutations detected in this study (Perez et al., 1997; Rivera et al., 1998; Desviat et al., 1999, 2001).
Based on our results, the first step of mutation screening for the population of Minas Gerais State should include the five most common mutations (V388M, R261Q, IVS10nt11, I65T, and R252W). This approach alone would allow the detection of 61% of the alleles and complete genotyping of 46% of the patients, with only five PCR and restriction digestion reactions/patient. Taken together, these results suggest that the mutation spectrum at the PAH locus of the population from Minas Gerais resembles more closely that from Portugal than those from São Paulo and southern Brazil states do. The PAH locus mutation spectrum in São Paulo and in southern Brazilian states clearly reflects the larger contingent of immigrants from middle and Eastern Europe. However, this hypothesis, which is in good agreement with historical information, needs to be confirmed by haplotyping studies. Most studies about the origins of Brazilian subpopulations are based on mitochondrial DNA or on Y-chromosome polymorphisms (Alves-Silva et al., 2000; Carvalho-Silva et al., 2001; Bortolini et al., 2003; Callegari-Jacques et al., 2003). The PAH gene offers a good alternative for evaluating the evolution of the autosomal genome. This gene apparently has no mutational or recombination hotspots, and it exhibits considerable haplotypic diversity. Both the haplotype origins and the extension of linkage disequilibrium of the locus are well characterized (Eisensmith and Woo, 1994; Kidd et al., 2000; Zschocke, 2003). Further haplotyping will allow us to establish PKU allele origins, as well as to make inferences on regional immigration events in this state. ACKNOWLEDGMENTS Special thanks go to the families that took part in this study. The authors thank the Núcleo de Pesquisa em Apoio ao Diagnóstico (NUPAD) for their collaboration in collecting patient’s samples. L.S. Lara and M. Castro Magalhães were supported by fellowships from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil. Research supported by grants from CAPES and NUPAD. We thank Roberto Giugliani, Maria Luiza Saraiva Pereira, and Angelina Xavier Acosta for their comments and suggestions. REFERENCES Acosta A, Silva Jr W, Carvalho T, Gomes M et al. (2001). Mutations of the phenylalanine hydroxylase (PAH) gene in Brazilian patients with phenylketonuria. Hum. Mutat. 17: 122-130. Alves-Silva J, da Silva Santos M, Guimaraes PE, Ferreira AC et al. (2000). The ancestry of Brazilian mtDNA lineages. Am. J. Hum. Genet. 67: 444-461. Benit P, Rey F, Blandin-Savoja F, Munnich A et al. (1999). The mutant genotype is the main determinant of the metabolic phenotype in phenylalanine hydroxylase deficiency. Mol. Genet. Metab. 68: 43-47. Bernegger C and Blau N (2002). High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: a study of 1,919 patients observed from 1988 to 2002. Mol. Genet. Metab. 77: 304-313. Blau N and Erlandsen H (2004). The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol. Genet. Metab. 82: 101-111. Bortolini MC, Salzano FM, Zago MA, Junior da Silva WA et al. (1997). Genetic variability in two Brazilian ethnic groups: a comparison of mitochondrial and protein data. Am. J. Phys. Anthropol. 103: 147-156. Bortolini MC, Salzano FM, Thomas MG, Stuart S et al. (2003). Y-chromosome evidence for differing ancient demographic histories in the Americas. Am. J. Hum. Genet. 73: 524-539. Callegari-Jacques SM, Grattapaglia D, Salzano FM, Salamoni SP et al. (2003). Historical genetics: spatiotemporal analysis of the formation of the Brazilian population. Am. J. Hum. Biol. 15: 824-834. Carreiro-Lewandowski E (2002). Newborn screening: an overview. Clin. Lab. Sci. 15: 229-238. Carvalho-Silva DR, Santos FR, Rocha J and Pena SD (2001). The phylogeography of Brazilian Y-chromosome lineages. Am. J. Hum. Genet. 68: 281-286. Cerone R, Schiaffino MC, Fantasia AR, Perfumo M et al. (2004). Long-term follow-up of a patient with mild tetrahydrobiopterin-responsive phenylketonuria. Mol. Genet. Metab. 81: 137-139. Desviat LR, Perez B, De Lucca M, Cornejo V et al. (1995). Evidence in Latin America of recurrence of V388M, a phenylketonuria mutation with high in vitro residual activity. Am. J. Hum. Genet. 57: 337-342. Desviat LR, Perez B, Gamez A, Sanchez A et al. (1999). Genetic and phenotypic aspects of phenylalanine hydroxylase deficiency in Spain: molecular survey by regions. Eur. J. Hum. Genet. 7: 386-392. Desviat LR, Perez B, Gutierrez E, Sanchez A et al. (2001). Molecular basis of phenylketonuria in Cuba. Hum. Mutat. 18: 252. Eiken HG, Odland E, Boman H, Skjelkvale L et al. (1991). Application of natural and amplification created restriction sites for the diagnosis of PKU mutations. Nucleic Acids Res. 19: 1427-1430. Eiken HG, Knappskog PM and Apold J (1993). Restriction enzyme-based assays for complete genotyping of phenylketonuria patients. Dev. Brain Dysfunct. 6: 53-59. Eisensmith RC and Woo SL (1992). Molecular basis of phenylketonuria and related hyperphenylalaninemias: mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum. Mutat. 1: 13-23. Eisensmith RC and Woo SL (1994). Population genetics of phenylketonuria. Acta Paediatr. (Suppl) 407: 19-26. Fisch RO (2000). Comments on diet and compliance in phenylketonuria. Eur. J. Pediatr. 159 (Suppl 2): S142-S144. Guldberg P, Rey F, Zschocke J, Romano V et al. (1998). A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am. J. Hum. Genet. 63: 71-79. Hofman KJ, Steel G, Kazazian HH and Valle D (1991). Phenylketonuria in U.S. blacks: molecular analysis of the phenylalanine hydroxylase gene. Am. J. Hum. Genet. 48: 791-798. Kidd JR, Pakstis AJ, Zhao H, Lu RB et al. (2000). Haplotypes and linkage disequilibrium at the phenylalanine hydroxylase locus, PAH, in a global representation of populations. Am. J. Hum. Genet. 66: 1882-1899. MacDonald A (2000). Diet and compliance in phenylketonuria. Eur. J. Pediatr. 159 (Suppl 2): S136-S141. McCabe LL, Therrell Jr BL and McCabe ER (2002). Newborn screening: rationale for a comprehensive, fully integrated public health system. Mol. Genet. Metab. 77: 267-273. Miller SA, Dykes DD and Polesky HF (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16: 1215. Perez B, Desviat LR, Die M, Cornejo V et al. (1993). Presence of the Mediterranean PKU mutation IVS10 in Latin America. Hum. Mol. Genet. 2: 1289-1290. Perez B, Desviat LR, De Lucca M, Schmidt B et al. (1996). Mutation analysis of phenylketonuria in South Brazil. Hum. Mutat. 8: 262-264. Perez B, Desviat LR and Ugarte M (1997). Analysis of the phenylalanine hydroxylase gene in the Spanish population: mutation profile and association with intragenic polymorphic markers. Am. J. Hum. Genet. 60: 95-102. Perez B, Desviat LR, De Lucca M, Cornejo V et al. (1999). Molecular characterization of phenylalanine hydroxylase deficiency in Chile. Mutations in brief No. 243. Online. Hum. Mutat. 13: 503. Pey AL and Martinez A (2005). The activity of wild-type and mutant phenylalanine hydroxylase and its regulation by phenylalanine and tetrahydrobiopterin at physiological and pathological concentrations: An isothermal titration calorimetry study. Mol. Genet. Metab. (in press). Rivera I, Leandro P, Lichter-Konecki U, Tavares de Almeida I et al. (1998). Population genetics of hyperphenylalaninaemia resulting from phenylalanine hydroxylase deficiency in Portugal. J. Med. Genet. 35: 301-304. Santana da Silva LC, Carvalho TS, da Silva FB, Morari L et al. (2003). Molecular characterization of phenylketonuria in South Brazil. Mol. Genet. Metab. 79: 17-24. Scriver CR and Kaufman S (2001). Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: The metabolic and molecular bases of inherited disease (Scriver CR, Beaudet A, Sly WS, Valle D et al., eds.). McGraw-Hill, New York, USA, pp. 1667-1724. Scriver CR, Hurtubise M, Konecki D, Phommarinh M et al. (2003). PAHdb 2003: what a locus-specific knowledgebase can do. Hum. Mutat. 21: 333-344. Serjeant GR (2000). Screening for sickle-cell disease in Brazil. Lancet 356: 168-169. Sokal RR and Rohlf J (1969). Analysis of frequencies. In: Biometry (Emerson R, Kennedy D, ParkBeadle RB and Whitaker GB, eds.). W.H. Freeman and Company, New York, USA, pp. 549-610. Steinfeld R, Kohlschutter A, Ullrich K and Lukacs Z (2004). Efficiency of long-term tetrahydrobiopterin monotherapy in phenylketonuria. J. Inherit. Metab. Dis. 27: 449-453. Yang Y, Drummond-Borg M and Garcia-Heras J (2001). Molecular analysis of phenylketonuria (PKU) in newborns from Texas. Hum. Mutat. 17: 523. Zschocke J (2003). Phenylketonuria mutations in Europe. Hum. Mutat. 21: 345-356. Zschocke J, Graham CA, Carson DJ and Nevin NC (1995). Phenylketonuria mutation analysis in Northern Ireland: a rapid stepwise approach. Am. J. Hum. Genet. 57: 1311-1317. |
|