![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
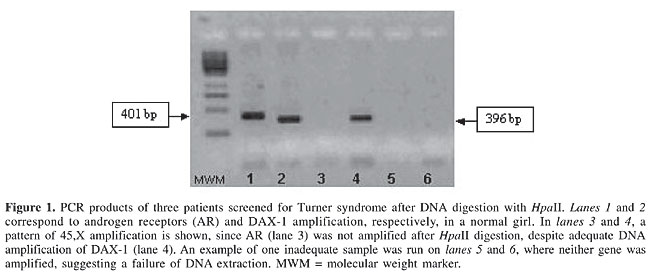
ABSTRACT. Turner syndrome (TS) is one of the most common human chromosomal abnormalities; it is characterized by the presence of one normal X chromosome and the complete or partial loss of the second X chromosome. The early recognition of TS patients allows for adequate therapy for short stature and pubertal sex steroid substitution. We developed a cost-effective molecular diagnostic tool that can be used to identify 45,X TS patients from dried blood spots, for possible use in neonatal screening for TS. We used a three-step method for 45,X TS detection: i) DNA extraction from dried blood spot samples, ii) pre-PCR HpaII digestion (methylation-sensitive enzyme) and iii) GeneScan analysis of selected cases. DAX-1 gene amplification was used to recognize DNA integrity, and the androgen receptor gene (Xq11-12), which is both a highly polymorphic and methylated gene, was used to determine the number of X chromosome alleles. Using this three-step diagnostic procedure, we detected apparent TS in 1/304 (0.33%) samples; such individuals should be submitted to clinical examination and karyotype confirmation. The three-step 45,X TS neonatal screening protocol is a simple, reliable, fast (under 30 h) and cost-effective diagnostic tool, useful for the neonatal detection of TS. Key words: Turner syndrome, Neonatal screening, Androgen receptor, Methylation INTRODUCTION Turner syndrome (TS) is one of the most common human chromosomal abnormalities; it is characterized by the presence of one normal X chromosome and the complete or partial loss of the second X chromosome. It is estimated to affect approximately 3% of all female fetuses, causing fetal death during the first trimester in 99% of the cases. Only 1% of these embryos survives to term (Lippe, 1991; Sybert and McCauley, 2004), accounting for an incidence of approximately 1:2500 to 1:5000 live female births (Gravholt et al., 1996). The karyotype 45,X is found in 40 to 60% of the detected cases (Álvarez-Nava et al., 2003). This syndrome is characterized by short stature, webbed neck, cardiac and renal abnormalities, as well as ophthalmologic and otologic disorders. Gonadal dysgenesis is associated with infertility in 98% of the cases. Autoimmune disorders, osteoporosis, carbohydrate intolerance, and arterial hypertension can also be present (Clement-Jones et al., 2000; Lowenstein et al., 2004). Only 20 to 30% of TS patients are diagnosed during the first year of life. The majority are recognized during childhood or adolescence. Unfortunately, almost 25% of the cases are not diagnosed until adulthood (Lespinasse et al., 1998; Conway, 2004). The early recognition of TS patients allows for adequate therapy for short stature and pubertal sex steroid substitution (Massa et al., 2005). Neonatal diagnosis of TS permits further investigation of associated anomalies, such as congenital heart and renal diseases, and tumor markers for dysgenic gonads. To date, no cost-effective methods are available for neonatal screening of TS patients; only karyotyping, pyrosequencing and multiple-marker genescans have been proposed. Molecular detection of 45,X TS patients could take advantage of the property that the second X chromosome is inactivated in blood and in most other tissues by methylation, in order to ensure expression of a single allele. At the same time, DNA methylation blocks DNA fragmentation by some restriction enzymes, such as HpaII. X chromosome methylation is absent in 46,XY or 45,X subjects; therefore, after HpaII digestion there is no amplification of genes spanning restriction sites on the X chromosome, such as the androgen receptor (AR) gene. On the other hand, subjects with two X chromosomes (46,XX) will have only one chromosome digested while the other one remains as a template for a subsequent PCR amplification. This molecular technique can use the small amounts of DNA that present in dried blood spots collected in routine neonatal screening programs. Our objective was to develop a cost-effective molecular diagnostic tool that can be used to identify 45,X patients from dried blood spots, potentially allowing for neonatal screening for 45,X TS. MATERIAL AND METHODS We analyzed DNA samples from dried blood spots collected by heel puncture of 304 phenotypically female newborns. Samples from patients with sexual ambiguity and card identification failure were excluded from this study. Informed consent was obtained from all parents according to the Helsinki Declaration, and the study was approved by the institutional Ethics Committee. Detection of the non-methylated allele by HpaII digestion DNA extraction and digestion were performed in a one-step reaction, employing the FTA Purification Reagent (cat# WB120204, FTA Whatman® International Ltd., Abington, Cambridge, UK) and the restriction enzyme HpaII (cat# ER512, MBI Fermentas). Briefly, a 7-mm punch from a previously spotted and dried FTA® Card (cat# WB 120208, Whatman® Bioscience Ltd., Abington, Cambridge, UK) was washed with FTA Purification Reagent. The samples were incubated with DNAzolTM (cat# 10503-027, InvitrogenTM Life Technologies, Carslbad, CA, USA) for 5 min, followed by 3 washes with TE-1 buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0). Digestion was performed directly from FTA discs in 50 µL, with 50 units of HpaII enzyme, enzyme-containing buffer and overnight incubation at 37°C. Digested DNA samples were submitted to PCR amplification, employing primers targeting the androgen receptor gene (OMIM 313700). The forward and reverse primers were: 5’ - GGG TAA GGG AAG TAG GTG GAA - 3’ and 5’ - ACT GCG GCT GTG AAG GTT - 3’ (InvitrogenTM). The PCR reaction included 20 pmol of each primer, 200 µmol of each deoxynucleotide, 0.5 U TaqDNA polymerase, 10X buffer, 50 mM MgCl2 (cat# N801-0055, GeneAmp - PCR reagent kit with AmpliTaq DNA polymerase, Perkin Elmer, Branchburg, NJ, USA) and H2O to obtain a final volume of 25 µL in a thermocycler (GeneAmp PCR System 9700 Applied Biosystem, Forster City, CA, USA). After an initial denaturation at 95°C for 5 min, the samples underwent 35 cycles of 94°C for 1 min, 65°C for 1 min and 72°C for 1 min and a final extension at 72°C for 10 min. The AR PCR product (401 bp) was analyzed by electrophoresis on 1.5% agarose gel containing ethidium bromide. DNA integrity was checked by co-amplification of a 396-bp fragment of the DAX1 gene (OMIM 300473), which has no restriction sites for HpaII, employing forward primer 5’ - GCT AGC AAA GGA CTC GGT - 3’ and reverse primer 5’ - CAG CTC TTT ATT CTT CCC TCA - 3’. HpaII cleaves only the non-methylated CCGG site, allowing DNA amplification only from the methylated AR allele. The expected result for normal girls (46,XX) is amplification of both AR and DAX-1, since there is a methylated allele. X chromosome monosomy (altered results) was seen as a single DAX-1 band with no AR gene amplification. Allele determination by the number of androgen receptor CAG repeats (GeneScan technique) DNA samples presenting altered results on HpaII restriction were subsequently re-extracted using PstI and evaluated by GeneScan analysis in order to determine the number of AR alleles; AR is a highly polymorphic gene, containing exon 1CAG repeats and is located at Xq11-12. The forward primer was labeled with 6-FAM (6-carboxyfluorescein): 5’ 6-FAM - GGG TAA GGG AAG TAG GTG GAA - 3’ and the non-labeled reverse primer was 5' - ACT GCG GCT GTG AAG GTT - 3'. The PCR included 5 pmol of the labeled sense primer, 20 pmol of non-labeled anti-sense primer, 200 µmol of each deoxynucleotide, 50 mM MgCl2, 10X buffer, 0.1 U Taq DNA polymerase (GeneAmp), and H2O to a final volume of 50 µL. PCR was run in a thermocycler GeneAmp PCR System 9700 (Applied Biosystems), under the following conditions: initial denaturation at 94°C for 3 min, followed by 30 cycles of 94°C for 1 min, 61°C for 1 min and 72°C for 1 min, with a final extension at 72°C for 7 min. The PCR product (395-bp fragment) was then submitted to capillary gel electrophoresis in an automated analyzer ABI PRISM 310 (Applied Biosystems), under the following conditions: 60°C, 7-9 µA° for 28 min for each sample. The results were analyzed employing the GeneScan software, which is able to determine the relative size of the amplified fragment; this varies according to the number of CAG repeats present in exon 1 of the AR gene. RESULTS Analysis of 304 samples gave normal results in 223 (73%), corresponding to AR and DAX-1 gene amplification, which excludes 45,X TS. DAX-1 gene amplification assures DNA integrity. AR gene amplification indicates the presence of a second, methylated, X chromosome that was not digested by HpaII enzyme (Figure 1).
The remaining 81 samples (27%), though they gave DAX-1 gene amplification, did not show a corresponding AR band; they were considered as possibly abnormal, requiring a second complete test. After this new extraction, digestion and PCR, 65/81 samples (80% of the repeat and 21% of the original samples) showed both DAX-1 and AR bands, being considered as normal results. However, 16/81 samples remained abnormal after the second test, which would mean a suspect rate of 5%. In order to decrease this high rate of suspect cases, a subsequent GeneScan analysis was performed on 15/16 samples, for which adequate DNA was available. The results from GeneScan were normal in 14 cases; two alleles were demonstrated, showing different numbers of CAG repeats. In only one case, a single allele size was detected, which could represent either a true X monosomy or two alleles of the same size. DISCUSSION Neonatal screening of TS could be a relevant procedure considering that it is a relatively frequent disease present in 1:2500 to 1:5000 newborn females. Early diagnosis would help in the recognition of complications, improve therapy effectiveness and could also permit the implementation of new methods for gonadal preservation. However, so far no cost-effective screening program is available for neonatal detection. Other groups have focused on prenatal detection of chromosomal abnormalities by using ultrasonography and multiple-marker screening (alpha-fetal-protein, hCG and estriol), which are not specific for TS detection (Wenstrom et al., 1994; Ruiz et al., 1999; Bronshtein et al., 2003; Falcon et al., 2005). We propose a new method for 45,X TS detection, using DNA extraction from dried blood spot samples, HpaII digestion pre-PCR (methylation-sensitive enzyme) and GeneScan analysis on selected suspect cases. Blood collection was performed on FTA cards, allowing easy transportation and adequate storage of samples. We were able to complete the whole testing process in less than 30 h, reporting results faster than the 5-7 days required for karyotyping. GeneScan analysis was even faster, with a turnaround time of under 24 h. Using the three-step diagnostic procedure, we found one case in 304 samples that should require patient recall for clinical examination and karyotype confirmation. This particular patient was not located for confirmation due to social problems faced by her family. This method was previously validated by our group for 45,X TS detection in karyotype-confirmed cases, using peripheral blood samples (Longui et al., 2002). Cost analysis of this method, using standard fees for technical staff in our country, demonstrated its viability as a screening test with a per-test cost of US$13.60. Molecular testing used in neonatal screening in our country is reimbursed at three times this cost and this, in turn, is also less expensive than karyotyping. An alternative approach would be to change the sequence of the three-step test, initiating with GeneScan analysis and then using HpaII digestion in non-informative cases (approximately 10%) (Racchi et al., 1998). This would decrease the cost and improve turnaround time but would require special equipment. It would be necessary to prospectively study the cost-benefit of using a single highly polymorphic AR marker or multiple markers, as proposed by other groups (Pereira et al., 2000). A limitation of this protocol is the ability to detect only 45,X TS patients and not other chromosomal abnormalities seen in these patients. However, this is the most frequent abnormal karyotype, accounting for almost 50% of the cases. Future attempts to improve the method should include the use of standard laboratory filter paper and improvement of digestion for more adequate and faster DNA extraction. At this point, the three-step protocol is already a suitable and cost-effective neonatal screening tool for 45,X TS patients. ACKNOWLEDGMENTS The authors thank FAPESP - Fundação de Amparo à Pesquisa do Estado de São Paulo and FAP - Fundação de Amparo à Pesquisa da Santa Casa de São Paulo (project # 03/307) for their generous financial support of this work. We are grateful to the Support Center for Scientific Publications of Santa Casa de São Paulo - Faculty of Medical Sciences for the editorial assistance. REFERENCES Álvarez-Nava F, Soto M, Sanchez MA, Fernandez E et al. (2003). Molecular analysis in Turner syndrome. J. Pediatr. 142: 336-340. Bronshtein M, Zimmer EZ and Blazer S (2003). A characteristic cluster of fetal sonographic markers that are predictive of fetal Turner syndrome in early pregnancy. Am. J. Obstet. Gynecol. 188: 1016-1020. Clement-Jones M, Schiller S, Rao E, Blaschke RJ et al. (2000). The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum. Mol. Genet. 9: 695-702. Conway GS (2004). Considerations for transition from paediatric to adult endocrinology: women with Turner’s syndrome. Growth Horm. IGF Res. 14 (Suppl A): S77-S84. Falcon O, Wegrzyn P, Faro C, Peralta CF et al. (2005). Gestational sac volume measured by three-dimensional ultrasound at 11 to 13 + 6 weeks of gestation: relation to chromosomal defects. Ultrasound Obstet. Gynecol. 25: 546-550. Gravholt CH, Juul S, Naeraa RW and Hansen J (1996). Prenatal and postnatal prevalence of Turner’s syndrome: a registry study. B.M.J. 312: 16-21. Lespinasse J, Gicquel C, Robert M and Le Bouc Y (1998). Phenotypic and genotypic variability in monozygotic triplets with Turner syndrome. Clin. Genet. 54: 56-59. Lippe B (1991). Turner syndrome. Endocrinol. Metab. Clin. North Am. 20: 121-152. Longui CA, Rocha MN, Martinho LC, Gomes GG et al. (2002). Molecular detection of XO - Turner syndrome. Genet. Mol. Res. 1: 266-270. Lowenstein EJ, Kim KH and Glick SA (2004). Turner’s syndrome in dermatology. J. Am. Acad. Dermatol. 50: 767-776. Massa G, Verlinde F, De Schepper J, Thomas M et al. (2005). Trends in age at diagnosis of Turner syndrome. Arch. Dis. Child. 90: 267-268. Pereira RW, Sturzeneker R and Pena SDJ (2000). Screening fetal losses for monosomy X with a simple PCR-based procedure. Genet. Mol. Biol. 23: 11-14. Racchi O, Mangerini R, Rapezzi D, Rolfo M et al. (1998). X chromosome inactivation patterns in normal females. Blood Cells Mol. Dis. 24: 439-447. Ruiz C, Lamm F and Hart PS (1999). Turner syndrome and multiple-marker screening. Clin. Chem. 45: 2259-2261. Sybert VP and McCauley E (2004). Turner’s syndrome. N. Engl. J. Med. 351: 1227-1238. Wenstrom KD, Williamson RA and Grant SS (1994). Detection of fetal Turner syndrome with multiple-marker screening. Am. J. Obstet. Gynecol. 170: 570-573. |
|