![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
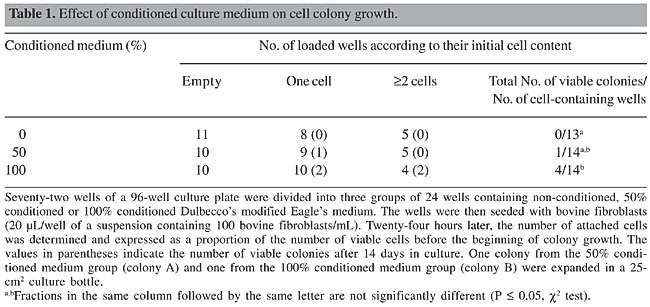
ABSTRACT. Transgenesis in cattle has provided numerous opportunities for livestock production. The development of nuclear transfer (NT) technology has improved the production of transgenic livestock. However, the isolation of pure colonies from a single transfection event remains laborious and can be a constraint in the production of transgenic livestock. We used 96-well cell culture plates to isolate cell lineages obtained from a single fibroblast transfected with the pCi-Neo plasmid. Since single mammalian cells do not grow well in fresh medium, we evaluated the use of conditioned medium. The neomycin phosphotransferase gene was detected in isolated colonies and NT embryos were produced from these cells. Multiplex-PCR assays were performed to detect the transfected fragment as well as autosomal satellite DNA in single NT embryos. This approach provided a reliable method for isolating transfected mammalian cells and for diagnosing the incorporation of desirable vectors in NT embryos. This method can reduce the time and cost of transgenic livestock production. Key words: Cattle, Cell isolation, Multiplex-PCR, Nuclear transfer, Transgene detection INTRODUCTION Transgenic technology allows the rapid introduction of new genes into animals to improve animal production, to express proteins with pharmaceutical properties, to generate models of genetic disease, and to study animal metabolism and the mechanisms controlling gene expression. The technique of microinjecting DNA into zygote pronuclei to produce transgenic livestock has been available for more than a decade (Pursel et al., 1989), but the low efficiency of transgene integration, the high cost of maintaining transgenic animals, and the long generation time are bottlenecks for the efficient use of this technology. Transgenic embryos produced by microinjection can be detected by PCR-based assays (Horvat et al., 1993). However, the high rates of false-positives caused by the contamination of DNA in the microinjection chambers, the presence of episomal plasmids, and the generation of chimeras are inherent difficulties of this technique (Krisher et al., 1994; Page et al., 1995). In contrast to microinjection, nuclear transfer (NT) of transgenic cattle cells provides precise gene targeting and allows the production of knock-in and knock-out animals, as well as a substantial increase in the proportion of live transgenic animals (Polejaeva and Campbell, 2000; Kuroiwa et al., 2002, 2004). However, cytoplasmic cleavage after NT and genome activation can produce non-transgenic, parthenogenetic structures that may be erroneously defined as blastocysts and transferred to the recipient animals. PCR-based detection methods are still the most appropriate choice for certifying the transgenic status of embryos produced by NT. Another limiting step in the production of transgenic livestock is the time required to isolate somatic cell clones, an important step for guaranteeing a single transgene-integration event. Somatic cell senescence and the possibility of colony contamination mean that an efficient protocol of cell isolation is fundamental for the successful production of transgenic animals. We developed a rapid method for isolating colonies grown from single transfected cells and combined this with the detection of the neomycin phosphotransferase gene (NeoR) in a single transgenic bovine embryo. The detection of the NeoR gene in biopsies of potentially transgenic NT embryos can reduce the time and cost of producing transgenic livestock and can minimize the wastage of animal resources. MATERIAL AND METHODS Cell culture and transfection Fibroblasts were isolated from bovine ear biopsies with three washes in 0.05% trypsin (Gibco), transferred to 25-cm2 cell culture bottles, and incubated in Dulbecco’s modified Eagle’s medium (DMEM, Gibco), supplemented with 10% fetal calf serum (Gibco), at 39°C in a 5% CO2 atmosphere (Freshney, 1994). After three passages, the fibroblast cultures were homogenous and the cells were transferred to 24-well culture dishes (105 cells/well) and grown for 24 h until they reached >70% confluency. The cells were then transfected with 0.5 µg of the pCi-Neo plasmid (Invitrogen) by lipofection with Lipofectin and Plus Reagent (Invitrogen), according to the manufacturer’s instructions. After 48 h, the culture medium was replaced by DMEM containing the G418 selective agent (500 µg/mL, Gibco) and grown for seven days. The surviving fibroblasts were detached and diluted (100 cells/mL) in fresh or conditioned medium and seeded in 96-well culture plates in 20-µL drops. DMEM was conditioned by growing non-transfected fibroblasts in 75-cm2 bottles at 39°C in 5% CO2 until they reached 80% confluency, at which point the medium was replaced and conditioned for 36 h. Cell attachment, growth and viability were assessed with an inverted microscope fitted with a heated platform to prevent thermal shock. Multiplication of isolated colonies Two strategies were used to expand the isolated colonies. In the first strategy, the confluent colonies were detached and the cells transferred to a 24-well culture dish together with the original non-transgenic fibroblasts in order to increase the cell density. After the first expansion in the 24-well dish, the cells were transferred to a 25-cm2 culture bottle and grown until they reached 80% confluency. The cells were then continuously selected by exposure to the antibiotic G418 (600 µg/mL) for 30 days in order to eliminate the non-transgenic cells. In the second strategy, the isolated colonies grown in 96-well plates were transferred individually to 24-well culture plates and maintained under G418 selection until they reached 80% confluency, at which point the cells were transferred to 25-cm2 culture bottles for the final growth phase. DNA extraction and PCR Fibroblast DNA was extracted from the contents of a 25-cm2 culture bottle by the salting-out procedure, with minor modifications (Miller et al., 1988; Biase et al., 2002). Fifty nanograms of genomic DNA was used in 25 µL of PCR mix (1 U Taq polymerase, 100 µM dNTP, 1 mM MgCl2, 5 pmol of each primer) and amplified 36 times using the following conditions: 93°C for 3 min, 93°C for 40 s, 58°C for 40 s, 72°C for 40 s, and 72°C for 5 min. The primers were designed to amplify a 410-bp fragment of the NeoR gene (sense: 5’-GAG-GCT-ATT-CGG-CTA-TGA-CTG-3’ and anti-sense: 5’-TCG-ACA-AGA-CCG-GCT-TCC-ATC-3’) and a 262-bp fragment of bovine satellite I DNA (Gaillard et al., 1981) (sense: 5’-AGG-TCG-CGA-GAT-TGG-TCG-CTA-GGT-CAT-GCA-3’ and anti-sense: 5’-AAG-ACC-TCG-AGA-GAC-CCT-CTT-CAA-CAC-GT-3’). DNA was extracted from single bovine embryos between the morula and blastocyst stages by treating the embryos with proteinase K or NP-40. Embryos cultured in vitro were washed twice in phosphate-buffered saline, drained and transferred individually to 0.2-mL PCR tubes, and the volume was completed to 8 µL with water. Subsequently, 2 µL of 5% or 10% NP-40 was added and the suspension was covered with two drops of mineral oil and subjected to four freeze-thaw cycles using liquid nitrogen and a thermo-block (95°C). A second set of embryos was transferred to 9 µL of 1X PCR buffer in 0.2-mL tubes, and 1 µl of proteinase K (2 or 20 mg/mL, Sigma) was added; the suspension was then covered with mineral oil and incubated at 55°C for 10 min. After digestion, the enzyme was heat inactivated (95°C, 5 min). Fifteen microliters of PCR mix was then added to the mechanical and enzymatic embryo lysates and the reactions were carried out as described above. The multiplex-PCR was done in two steps: in the first step, only the NeoR primers were used in 27 cycles of PCR. Subsequently, 1 µL of the first PCR mixture was added to a second PCR reaction containing the NeoR primers and the satellite I specific primers, and the reactions were carried out for 33 extra cycles. Nucleus transfer embryos production and culture To produce transgenic and non-transgenic embryos, cells from transgenic colonies and non-transgenic parental cells were used as nucleus donors. The cells were grown in 25-cm2 culture bottles until two days after they reached 100% confluency, at which point they were treated with 0.05% trypsin and washed twice in 1 mL TCM 199 medium (Gibco). The NT technique was carried out with high quality oocytes (grades 1-2, according to the IETS classification in Stringfellow and Seidel, 1998) selected from ovaries of slaughtered cows. Cumulus oocyte complexes were maturated in vitro in TCM 199, supplemented with 10% fetal calf serum, 24 U/mL luteinizing hormone and 10 µg/mL follicle-stimulating hormone for 22 h. The mature oocytes were enucleated with a micromanipulator (Narishige 188) attached to an inverted microscope (Nikon TDM) and the nucleus donors were placed in the perivitelinic space before electrofusion (Wilmut et al., 1997). The oocyte-derived cytoplast and the fibroblast were fused by electrofusion (BTX equipment) in two pulses of 2.1 Kv/cm for 30 µS. After fusion, the reconstructed structures were activated with ionomycin for 5 h, followed by 6-dimethylaminopurine (Sigma) for a further 5 h. The NT embryos were cultured over a monolayer of cumulus cells in SOF medium (Holm et al., 1999) at 39°C in a 5% CO2 atmosphere for 6-7 days, until they reached the compacted morula or blastocyst stage (Holm et al., 1999; Dode et al., 2002). RESULTS Effect of conditioned medium on transfected colony growth Of 72 wells seeded with cell-suspension droplets, 31 contained no cells, 27 contained one cell and 14 contained two or more cells (Table 1). The 41 cell-containing wells were divided into three groups according to the composition of the culture medium: 13 colonies were grown in non-conditioned medium, 14 colonies were grown in 50% conditioned medium and 14 colonies in 100% conditioned medium. After 14 days in culture, no colony grew in non-conditioned medium, one colony grew in 50% conditioned medium and four grew in 100% conditioned medium. Of the five isolated colonies, two (identified as A and B), grew more and were used in the colony-expansion experiment. Based on these results, only colonies obtained with fully conditioned medium were used in all subsequent colony-isolation assays.
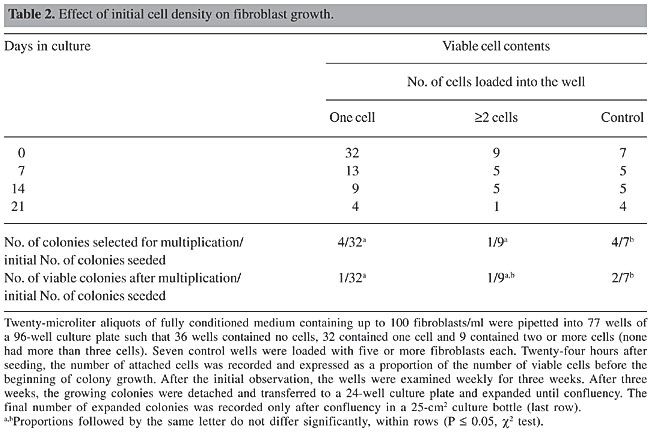
Isolation and multiplication of transfected fibroblasts In a second assay, we tested the efficiency of fully conditioned medium for supporting single-cell growth in 96-well plates. Of 77 wells seeded with cell-suspension drops, 36 wells contained no cells, 32 contained one cell, and 9 contained two or more cells (Table 2). As a control for cell density, more than 5 cells/well were seeded in eight wells. After 20 days, four colonies were isolated from the one-cell group, one from the group with ³2 cells (11%), and four from the control group. Only rarely were more than three cells found in the group with ³2 cells. Only one colony, from the one-cell group, grew to confluency in a culture bottle. The colony from the group with ³2 cells reached confluency when transferred to a culture bottle, as did two colonies from the control group. There was a significant difference in the percentage of confluent colonies in the control wells compared to the colonies initially formed by one or two cells. Therefore, the initial cell density in the culture drop affects the subsequent sustainability of colony growth and expansion. The remaining colonies were monitored throughout the entire assay period, but none of them reached confluency. To circumvent the negative effect of a low cell density on colony expansion, we mixed the transgenic cells of colonies A and B from the first assay (Table 1) with their non-transgenic counterparts in order to improve the initial growth. Using this co-culture assay, we were able to expand the original colonies in 25-cm2 culture bottles to reach confluency in two weeks.
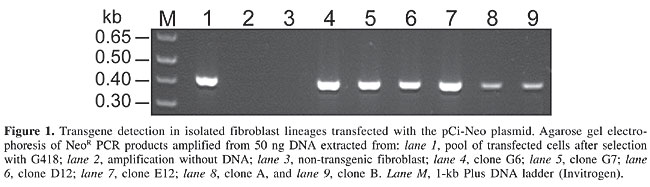
Transgene detection in cells and embryos The DNA of confluent cultures (in 25-cm2 culture bottles) was extracted and assayed by PCR in order to amplify the NeoR gene (Figure 1). The original, non-isolated transfected cells (Mosaic) contained the NeoR gene (positive control), as did the colonies that were expanded without co-culture with non-transfected fibroblasts. In contrast, the expanded transfected lineages co-cultured with non-transgenic cells showed only a slight amplification of the NeoR band (Figure 1, lanes 8 and 9), which suggested contamination with non-transgenic cells, despite the selection pressure produced by the antibiotic. For this reason, the co-culture of transgenic and non-transgenic fibroblasts was abandoned in the subsequent colony-isolation assays.
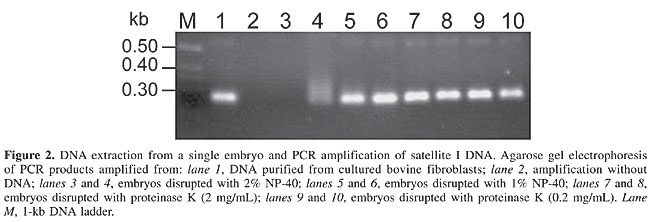
We observed an effect of the embryo lysis protocol on the outcome of the standard PCR assays (Figure 2). Both the enzymatic and the physical methods yielded embryonic DNA suitable for amplification. However, in samples treated with 2% NP-40, the PCR reaction was inhibited. Hence, a buffer containing 1% NP-40 was used in all subsequent experiments.
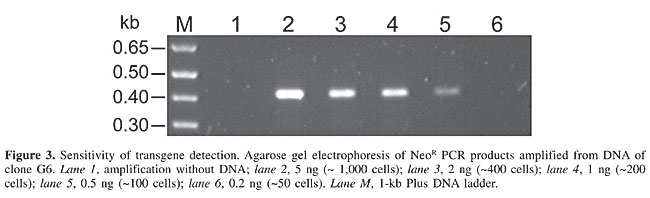
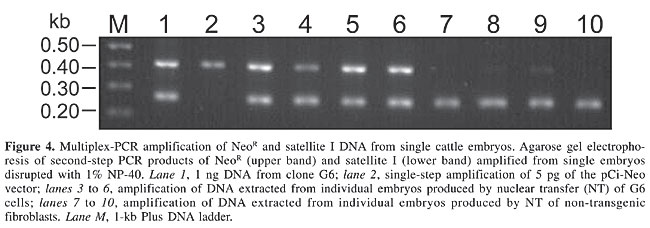
To determine the sensitivity of our PCR, the NeoR gene was amplified in decreasing concentrations (5, 2, 1, 0.5, and 0.2 hg) of transgenic DNA isolated from transgenic cells. The construct fragment was detected in samples containing 0.5-1 ng DNA (Figure 3). This quantity corresponded to ~100-150 cells, assuming that one cell contained 6.6 pg genomic DNA (Lewin, 1994); consequently, we decided that a two-step PCR would be necessary to detect the transfected fragment in cattle embryos. Four non-transgenic and four transgenic embryos were used as individual templates in a two-step multiplex PCR. After the first amplification, a slight band corresponding to the transgene (NeoR gene) was detected in only two samples (data not shown). In the second step, 1 µL of the first reaction was used to co-amplify the satellite repetitive region together with the NeoR gene. As expected, the bovine satellite region was amplified in all of the embryos, but only the embryos derived from transgenic fibroblasts amplified the NeoR gene (Figure 4).
DISCUSSION Transgenic technology in livestock is a powerful tool to produce models to study human diseases and for obtaining proteins of pharmacological interest (Pursel et al., 1989; Stice et al., 1998; Brink et al., 2000). These possible applications expanded considerably with the advent of NT technology, which provided more efficient means for producing viable and stable transgenic embryos, when compared with pronuclear injection (Brink et al., 2000; Polejaeva and Campbell, 2000; Montoliu, 2002). However, the NT procedures are still inefficient and expensive, particularly because of the low rate of certified transgenic embryos that are produced and the high losses during pregnancy and among neonates (Schnieke et al., 1997; Stice et al., 1998). One possible explanation for the high rate of abortion of transgenic blastocysts, compared to non-transgenic blastocysts, is the extensive culturing and manipulation in vitro that is necessary to transfect, select and isolate certified transgenic nuclear donor cells. Extensive manipulation can prevent correct gene expression patterns during embryonic development (Farin et al., 2001; Young et al., 2001; Bertolini et al., 2002). In addition, working with a specific integration event, it is necessary to ensure that the transgenic nuclear donors are derived from a single transfected cell. Hence, the isolation of cell clones is essential for the efficient production of transgenic embryos. We have demonstrated a strategy to transfect, select, isolate, multiply, and detect transgenic nuclear donor cells from cattle and, after transfer, detect the NeoR marker gene in the transgenic embryos. Some of the articles recently published on this subject lack details on the methods that allow transgenic cattle production, particularly on how the transgenic clonal lines are isolated and multiplied (Cibelli et al., 1998; van Berkel et al., 2002; Kuroiwa et al., 2002, 2004). One method frequently employed to isolate colonies from a single transfection event is the use of cloning rings (Freshney, 1994). However, this method does not guarantee the isolation of certifiable clonal parental cells, and it is prone to contamination by grease and ceramic or steel ring manipulations. This obstacle makes difficult the manipulation of several colonies at the same time, and since the growth capacity of isolated colonies is limited, it limits the number of nuclear donors that can be obtained. Our method, based on 96-well culture plates, allows one to individually manipulate several transfected colonies at the same time. After growth for 6-7 weeks, we were able to isolate and multiply up to 106 fibroblasts from a single transgenic cell, all of which could be used as nuclear donors to produce embryos harboring the expected introduced fragment. This process was made possible by starting clonal colonies from 96-well culture plates, many of which originally contained only one transfected cell. A solid substrate, cytokines and growth factors are necessary for mammalian fibroblasts to grow in vitro (Takehara, 2000). The essential growth factors secreted by fibroblasts and needed for their proper growth in culture include epidermal growth factor, platelet-derived growth factor, basic fibroblast growth factor (b-FGF), transforming growth factor-b (TGF-b), and connective tissue growth factor (CTGF) (Rubin et al., 1989; Takehara, 2000). To provide all of these factors, we used conditioned culture medium obtained from non-transgenic fibroblast cell cultures. The efficiency of this strategy was evident from the deficient growth seen in non-conditioned medium, which showed that conditioned medium is essential for adequate growth, probably due to the autocrine stimulatory effect of TGF-b and CTGF secreted by non-transfected fibroblasts. In order to estimate the rate of transgenic production, we focused on transgene fragment detection in a single preimplantation embryo. Cells of clonal origin were submitted to PCR to certify their exogenous gene incorporation status, before the NT assays. Several blastocysts were produced by NT, using transgenic fibroblasts as nuclear donors (data not shown). Four blastocysts, with good morphological aspect, were isolated and disrupted to detect the NeoR gene and satellite I DNA by PCR. Previous experiments from our laboratory showed that, in many cases, a single PCR round is not enough to detect the introduced fragment. This may be due to the fact that the cells of a cattle blastocyst number less than 150. Therefore, in order to increase the chance of detection, we applied two rounds of PCR amplification. This two-step PCR allows transgenic and non-transgenic sample identification with 100% efficiency (Figure 4). It is known that electric pulses and manipulation result in the mobilization of intracytoplasmatic calcium reservoirs, during the NT, and can activate blastomere cleavage in several reconstructed cytoplasts, producing parthenogenetic embryos (Meo et al., 2004). As a result, activated cytoplasts can develop parthenogenetically to produce blastocyst-like structures that may erroneously be considered to be blastocysts, and the transfer of these pseudoblastocyts to recipient animals can result in inviable pregnancies. By using the strategy to detect the transgene in trophoblast biopsies of NT blastocysts, the possibility of transferring parthenogenetic structures to recipient heifers can be minimized or even eliminated. The viability of transgenic embryos depends on the nature of the inserted construct and the place of insertion in the host genome. We have shown that it is possible to quickly detect and measure the production rate of embryos harboring the desirable construct. Based on the results of PCR analyses, pregnancies of negative transgenic embryos can be interrupted, thereby minimizing the waste of animal resources. Such an approach should result in a more economical production of transgenic livestock. ACKNOWLEDGMENTS We thank Dr. Eugen S. Gander and Dr. Carlos J.H. Souza for helpful discussions and for assistance in preparing the manuscript. Research supported by EMBRAPA and PRODETAB. REFERENCES Bertolini M, Beam SW, Shim H, Bertolini LR et al. (2002). Growth, development, and gene expression by in vivo- and in vitro-produced day 7 and 16 bovine embryos. Mol. Reprod. Dev. 63: 318-328. Biase FH, Franco MM, Goulart LR and Antunes RC (2002). Protocol for extraction of genomic DNA from swine solid tissues. Genet. Mol. Biol. 25: 313-315. Brink MF, Bishop MD and Pieper FR (2000). Developing efficient strategies for the generation of transgenic cattle which produce biopharmaceuticals in milk. Theriogenology 53: 139-148. Cibelli JB, Stice SL, Golueke PJ, Kane JJ et al. (1998). Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 280: 1256-1258. Dode MAN, Mattos L and Rumpf R (2002). In vitro production of embryos in SOF medium under high oxygen tension. In: Annual Conference of the International Embryo Transfer Society, Foz do Iguaçu, 2002. Theriogenology, 2002, Vol. 57, p. 661. Farin PW, Crosier AE and Farin CE (2001). Influence of in vitro systems on embryo survival and fetal development in cattle. Theriogenology 55: 151-170. Freshney RI (1994). Culture of animal cells: a manual of basic technique. Wiley-Liss, New York, NY, USA. Gaillard C, Doly J, Cortadas J and Bernardi G (1981). The primary structure of bovine satellite 1.715. Nucleic Acids Res. 9: 6069-6082. Holm P, Booth PJ, Schmidt MH, Greve T et al. (1999). High bovine blastocyst development in a static in vitro production system using SOFaa medium supplemented with sodium citrate and myo-inositol with or without serum-proteins. Theriogenology 52: 683-700. Horvat S, Medrano JF, Behboodi E, Anderson GB et al. (1993). Sexing and detection of gene construct in microinjected bovine blastocysts using the polymerase chain reaction. Transgenic Res. 2: 134-140. Krisher RL, Gibbons JR, Canseco RS, Johnson JL et al. (1994). Influence of time of gene microinjection on development and DNA detection frequency in bovine embryos. Transgenic Res. 3: 226-231. Kuroiwa Y, Kasinathan P, Choi YJ, Naeem R et al. (2002). Cloned transchromosomic calves producing human immunoglobulin. Nat. Biotechnol. 20: 889-894. Kuroiwa Y, Kasinathan P, Matsushita H, Sathiyaselan J et al. (2004). Sequential targeting of the genes encoding immunoglobulin-micro and prion protein in cattle. Nat. Genet. 36: 775-780. Lewin B (1994). Genes V. Oxford University Press, Oxford, New York, USA. Meo SC, Leal CL and Garcia JM (2004). Activation and early parthenogenesis of bovine oocytes treated with ethanol and strontium. Anim. Reprod. Sci. 81: 35-46. Montoliu L (2002). Gene transfer strategies in animal transgenesis. Cloning Stem Cells 4: 39-46. Page RL, Canseco RS, Russell CG, Johnson JL et al. (1995). Transgene detection during early murine embryonic development after pronuclear microinjection. Transgenic Res. 4: 12-17. Polejaeva IA and Campbell KH (2000). New advances in somatic cell nuclear transfer: application in transgenesis. Theriogenology 53: 117-126. Pursel VG, Pinkert CA, Miller KF, Bolt DJ et al. (1989). Genetic engineering of livestock. Science 244: 1281-1288. Rubin JS, Osada H, Finch PW, Taylor WG et al. (1989). Purification and characterization of a newly identified growth factor specific for epithelial cells. Proc. Natl. Acad. Sci. USA 86: 802-806. Schnieke AE, Kind AJ, Ritchie WA, Mycock K et al. (1997). Human factor IX transgenic sheep produced by transfer of nuclei from transfected fetal fibroblasts. Science 278: 2130-2133. Stice SL, Robl JM, Ponce de Leon FA, Jerry J et al. (1998). Cloning: new breakthroughs leading to commercial opportunities. Theriogenology 49: 129-138. Stringfellow AD and Seidel MS (1998). Manual of the International Embryo Transfer Society. International Embryo Transfer Society, Savoy, IL, USA. Takehara K (2000). Growth regulation of skin fibroblasts. J. Dermatol. Sci. 24: S70-S77. van Berkel PH, Welling MM, Geerts M, van Veen HA et al. (2002). Large scale production of recombinant human lactoferrin in the milk of transgenic cows. Nat. Biotechnol. 20: 484-487. Wilmut I, Schnieke AE, McWhir J, Kind AJ et al. (1997). Viable offspring derived from fetal and adult mammalian cells. Nature 385: 810-813. Young LE, Fernandes K, McEvoy TG, Butterwith SC et al. (2001). Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat. Genet. 27: 153-154. |
|