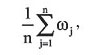![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
ABSTRACT. The coat protein (CP) of the family Luteoviridae is directly associated with the success of infection. It participates in various steps of the virus life cycle, such as virion assembly, stability, systemic infection, and transmission. Despite its importance, extensive studies on the molecular evolution of this protein are lacking. In the present study, we investigate the action of differential selective forces on the CP coding region using maximum likelihood methods. We found that the protein is subjected to heterogeneous selective pressures and some sites may be evolving near neutrality. Based on the proposed 3-D model of the CP S-domain, we showed that nearly neutral sites are predominantly located in the region of the protein that faces the interior of the capsid, in close contact with the viral RNA, while highly conserved sites are mainly part of b-strands, in the protein’s major framework. Key words: Neutral evolution, Maximum likelihood, PAML INTRODUCTION Luteoviruses have been reported on potato plantations for a long time, and they have also been seen in several other crops worldwide (Oswald and Houston, 1951; McKee, 1964; Harrison, 1999). However, it was not until the last decade that researchers attempted to group them into a single taxon, the family Luteoviridae (D’Arcy and Mayo, 1997). Viruses of the family Luteoviridae are phloem-limited and aphid transmitted in a circulative, non-propagative manner. Their genetic material is composed of a single, positive sense, non-polyadenylated RNA (Mayo and Ziegler-Graff, 1996). These viruses have a complex biology and are considered one of the most ecologically successful and economically important groups of plant viruses (Harrison, 1999). The family Luteoviridae has been divided into three genera based on differences in the RdRp and structural proteins: Luteovirus, Polerovirus and Enamovirus. On average, their genomes are 5700 nucleotides long and exhibit two major portions separated by an intergenic region of 100-200 nucleotides (Mayo and Ziegler-Graff, 1996). The nonstructural sequences located at the 5’ portion of the genome are highly variable among luteoviruses (Habili and Symons, 1989), where as the 3’ portion of the genome encloses a conserved structural coat protein (CP), CP-readthrough sequences and the P17 movement protein. The virion in the family Luteoviridae is an icosahedral structure with a diameter of 25 nm and probably consists of as much as 180 subunits of the CP organized in T = 3 symmetry (Harrison, 1999). The CP is considered to possess two main domains, the R region, situated at the N-terminal of the protein, and an S region, which comprises the structure’s major framework (Terradot et al., 2001). 3-D models obtained by homology depicted conserved secondary and tertiary structures, such as the jellyroll shape, present in plant and animal viruses (Dolja and Koonin, 1991; Mayo and Ziegler-Graff, 1996; Terradot et al., 2001; Brault et al., 2003, Lee et al., 2005). Studies using point mutations have elucidated the involvement of the CP in several stages of the viral life cycle. The protein is critical to the virus association with the aphid vector and may interact with cell receptors in the accessory salivary glands of the organism (Gray and Gildow, 2003). Therefore, CP is directly related to the specificity and the rate of viral transmission to the host plant. Also, this protein participates in several post-transmission stages, such as particle packaging and viral accumulation within the plant (Reutenauer et al., 1993; Peiffer et al., 1997; Brault et al., 2003). Despite its importance, extensive studies on the molecular evolution of the Luteoviridae CP are lacking. Recently, Guyader and Ducray (2002) analyzed 19 sequences of the Potato leafroll virus and reported that the overlapping open reading frames 3 and 4 are subject to differential selective pressures. This result indicates that some sites might have undergone relaxation of evolutionary constraints and that positive selection could have shaped the overall P3 sequence diversity found within the family. In the present study, we investigate the heterogeneity of selective forces acting on sites of the coat protein of luteoviruses as well as the potential differences in nonsynonymous rate evolution between the genera Polerovirus and Luteovirus. Our analyses show that the CP was subjected to significant differential selection and it is statistically reasonable to assume neutral evolution on many codon sites. Finally, we compare our results with the proposed 3-D models in the literature to envisage the biological meaning of such heterogeneity. MATERIAL AND METHODS Complete sequences of the CP coding region were retrieved from the GenBank (accession numbers shown in Table 1). A total of 48 sequences were obtained and then divided into two groups for later analyses. The first group consisted of the full sample of the 48 sequences (large data set), while the second was a reduced sample of 17 sequences retrieved from all genomes of Luteoviridae available to date (small data set). This was done to test the sensitivity of the methods used for taxon sampling, since it has already been argued that the number of sequences analyzed may influence the evolutionary parameters inferred in this work (Yang, 1998).
When collecting the sequences, we have favored the sampling of the most taxonomically diverse set to gather an accurate picture of Luteoviridae genetic variability. We have also avoided the usage of several highly similar sequences, since they would not increase the robustness of our estimates. All alignments were performed by Clustal W (Thompson et al., 1994). Gaps and ambiguous characters were eliminated, resulting in 179 analyzable codon sites. The phylogenies of the sequences studied were constructed in PAUP 4.b10 using the maximum likelihood method and the HKY + G model (Hasegawa et al., 1985). Model choice and parametric estimates were obtained in MODELTEST 3.5 using the likelihood ratio test with the significance level set at 1% (Posada and Crandall, 1998). The substitution model parameters estimated for the small data set were: freqA = 0.28, freqC = 0.27, freqG = 0.25, freqT = 0.19; Ts/Tv = 1.0; a = 0.89. For the large data set, the parameters were basically the same: freqA = 0.29, freqC = 0.28, freqG = 0.24, freqT = 0.19; Ts/Tv = 1.14; a = 0.86. Maximum likelihood analyses were performed on both data sets to verify the occurrence of differential selective pressures acting on sites of the coding sequence. The method of Yang et al. (2000), also available in PAML 3.13 (Yang, 1997), was used with five models of w evolution: the neutral (M1), selection (M2), discrete (M3), beta (M7), and beta&w (M8). We also compared these results with the recently proposed modifications of the models of Yang et al. (2000): the NearlyNeutral (M1a), PositiveSelection (M2a) and Beta&w > 1 (Wong et al., 2004, available in PAML 3.14). Tests for positive selection can be carried out by comparing the log-likelihoods of the M1-M2 and M7-M8 models in PAML 3.13 and the modified M1a-M2a and M7-M8 and w > 1 models in PAML 3.14. Finally, the comparison of the M3 model against the M2 or M2a is expected to provide a general picture of the heterogeneity of w values along the codons, although the hypothesis of positive selection is not really tested (Wong et al., 2004). Positive selection analysis of data sets with several sequences is time consuming. Therefore, to calculate site-specific w’s on the large data set, we fixed the branch lengths at the values inferred by the PAML’s M0 model. This approach eliminates the necessity of estimating 94 extra-parameters for each run and, technically, does not largely influence the overall results, since site-specific models should estimate the same branch length values inferred by the M0. All analyses were triple-checked by inputting different initial w’s (0.1, 0.5 and 1.5) to avoid parametric estimates from local optima. Biologically, it is meaningful to analyze our estimates in light of what is known of the 3-D structure of the CP. Terradot et al. (2001), Brault et al. (2003) and Lee et al. (2005) have proposed 3-D models of the S-domain of this protein based on homologous comparisons with related proteins found in virus families close to the Luteoviridae. This domain is fundamental for the virus biology since it forms the major structure of the capsid. Therefore, we projected the site-specific w values inferred to the Potato leafroll virus PDB file of Terradot et al. (2001) to envisage the association of selective pressure and protein structure. To calculate site specific w’s, we used the average posterior w values for each codon position under the M3 model (with K = 4 classes). For a given codon position i, the average posterior wi under the M3 equals the sum of the terms wK pK over all K = 4 classes of the model, w1 p1 + w2 p2 + w3 p3 + w4 p4, where pK is the Bayesian posterior probability of wK. These values can be obtained from the rst file, which is written by PAML after each run. We have also calculated average posterior w’s for a given region of the gene/protein by averaging the posterior averages of the codons in that region. In this case, if the region has n sites, its average
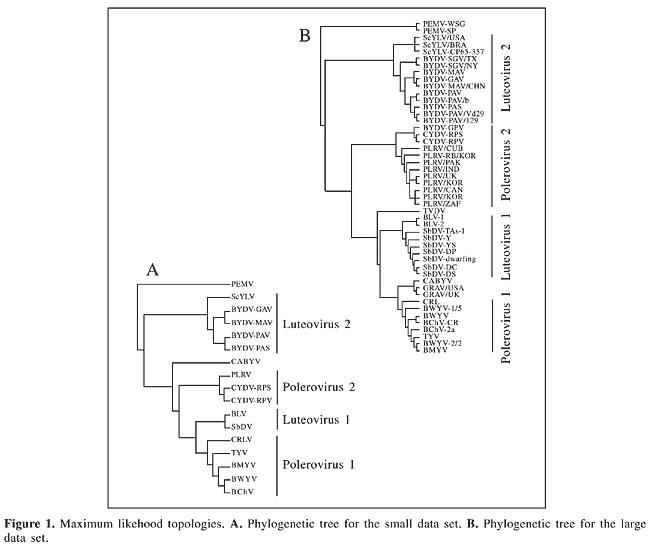
where wj is calculated exactly as wi above. RESULTS AND DISCUSSION Phylogenetic trees The maximum likelihood phylogenies obtained by our analyses did not recover the traditional taxonomic groups considered in the literature (Figure 1). The Luteovirus species BLRV and SbDV were included in the genus Polerovirus, while ScYLV, an unclassified virus with polerovirus affinities, was found within the genus Luteovirus. This result is expected to hold since it has already been demonstrated that the CP coding region has undergone recombination events during Luteoviridae evolution (Habili and Symons, 1989). Classification at the species level in the family is carried out by means of replication strategies and BLRV and SbDV polymerases are similar to those found in members of the genus Luteovirus, although their structural proteins are close to the genus Polerovirus (Rathjen et al., 1994; Domier et al., 2002). The inverse is true for ScYLV (Maia et al., 2000; Moonan et al., 2000; Smith et al., 2000).
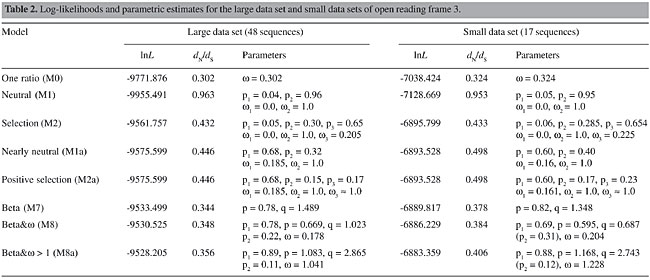
The phylogenetic position of the GRAV has never been thoroughly investigated, but the alignment provided by Scott et al. (1996) clearly indicates its kinship with the CABYV (75% similarity) and, therefore, justifies its inclusion within the genus Polerovirus. In our trees, the position of the CABYV is not fully resolved, since in the small data set it is a sister group of the [Polerovirus 2 (Luteovirus 1, Polerovirus 1)] group and in the large data set it becomes the outgroup of one of the Polerovirus clades (Polerovirus 1). Although each of the groups defined in Figure 1 are supported by bootstrap values higher than 80%, the basal nodes that disrupt the Luteovirus and Polerovirus genera are not statistically significant and, hence, much of the incongruences found between our trees and traditional taxonomic schemes may be also due to phylogenetic error. Such a problem, however, did not influence the estimation of site-specific parameters, since positive selection inference relies mainly on the overall sequence diversity (Yang et al., 2000). For instance, Yang and co-workers (2001) reported that even a star tree rendered similar parametric estimates when compared to the maximum likelihood topology of the vertebrate b-globin gene. Selective pressures acting on codon sites Codon-based analyses demonstrated that there were no drastic differences between the large and the small data sets (Tables 2 and 3), suggesting that parametric estimates, in this case, are robust to taxon sampling. Therefore, we will discuss on the large data set estimates, except when stated otherwise. Both data sets are best explained when models that incorporate neutral or positive selection categories are considered. Moreover, the discrete model, which depicts the heterogeneity of selective forces on the coding region, needs four categories to fit the data. Model comparisons that test for positive selection showed that it is not reasonable to assume it (Table 2). When comparing the M1-M2 models, the third category inferred by the M2 presented w < 0, and therefore, although this model is significantly better than M1, no positively selected sites were inferred. However, it is clear that a large number of sites are evolving at w values close to neutrality and that the assumption of negative selection at w = 0 is not realistic for the data. The neutral model M1 estimates 96% of the sites to be evolving at w = 1, this number falls to 30% when the w = 0.205 category is created in M2, which indicates that sites under 0 < w < 1 are indeed associated with the neutral category. Since the percentages of sites calculated to be under w = 0 are statistically identical, there is little doubt that the excess of neutral sites in M1 is due to inappropriate categorical association.
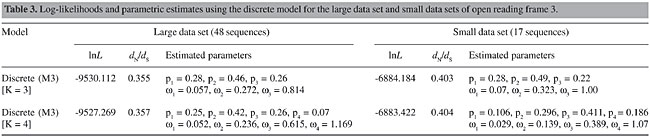
Theoretically, the comparison between M1a and M2a is more adequate, since there is no constraint on the negative selection category of M1a, and the M2a model is forced to infer sites with w > 1, resulting on a proper test of positive selection (Wong et al., 2004). Therefore, if there is a small number of sites with w > 1, they are expected to be assigned to this positive selection category with naive empirical Bayes (NEB) posterior probabilities >95%. Our estimates corroborate this theoretical prediction and restate that a significant number of codons in the coat protein are evolving near neutrality. The negative selection category of M1a is estimated at w = 0.185 and accounts for 68% of the sites, while the percentage of neutral sites is 32%. Note that these values are very close to the M2a estimates. The forced positive selection category of the M2a model was estimated at w » 1, accounting for 17% of the sites. This value is indistinguishable from neutrality and, in fact, the LRT statistic between M1a and M2a is null. Therefore, positive selection is rejected. As in M1a, M2a also infers that 68% of the sites are evolving at w near 0.2 and that about 32% of the sites are neutral (the sum of p2 and p3). Tests with models that use the beta distribution followed the same pattern recovered by the neutral-selection models. The M8 was not significantly best fit to the data when compared against the M7 (c2 = 5.95, d.f. = 2). Moreover, the inferred w is <1, which eliminates the possibility of positive selection. The M7-M8a comparison is similar to the M7-M8 except for the constraint imposed on the inferred w to be >1 in M8a. Once this model forces the existence of positive selected sites, it is predictable to find such sites. However, although M8a fits the data better than does M7 (c2 = 10.59, d.f. = 2), none of the w > 1 sites shows NEB posterior probability >95%, and moreover, the positive selection class was estimated near neutrality at w = 1.041. Diversifying selection then cannot be securely assumed. While testing the heterogeneity of selective forces along the open reading frame, the log-likelihood of the discrete model M3 with three classes (K = 3) was significantly better than the M2 and M2a. Moreover, the discrete model with four classes (K = 4) fits the data better when compared with K = 3; the addition of a fifth class did not significantly augmented the log-likelihood of the model. Thus, M3 (K = 4) best describes the variation of w values on CP. One of the classes of this model shows w > 1; this value, however, is also quite close to neutrality (w = 1.169) and, again, positive selection is weakly upheld (Table 3).
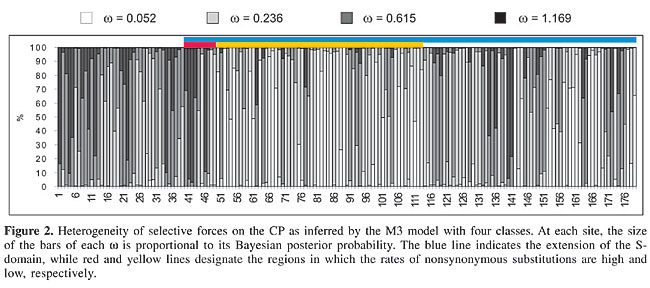
It is worth noting that the diversity found in site-specific w values was not randomly distributed along the coding region (Figure 2). The first 48 codons of the alignment, in the R region of the protein, account for the majority of the sites with w values higher than or close to one, while the middle region includes most of the sites under strong negative selection. When the estimates obtained from the M3 (K = 4) model are averaged, this separation is clearly illustrated. The R region sites possess an average
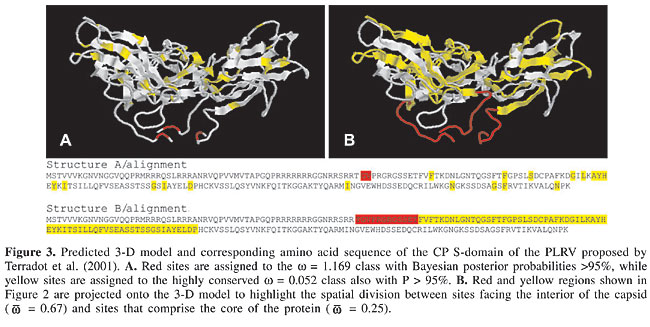
A considerable number of sites were estimated to be neutrally evolving by the neutral-selection models. This is particularly evident when the M2 is considered, since the estimated w = 0.205 class accounted for 65% of the sites, while 30% were assigned to the neutral class. Since the M1, M1a, M2, and M2a models force the w = 1 (neutral) category, it remains unclear whether the neutrality hypothesis is correct for these sites. A test for neutrality in this case would be the comparison of the M2-M3 (K = 3) or the M2a-M3 (K = 3) models. The M3 model with three classes estimates a third w that is not forced to be =1 as in M2 and M2a. Therefore, if this model fits the data better than does M2 or M2a and none of the estimated w’s in M3 is equal or close to one, the occurrence of neutral sites is not statistically supported. Indeed, as already stated, the M3 (K = 3) best fits the data when tested against M2 (c2 = 63.29, d.f. = 1) and M2a (c2 = 90.97, d.f. = 1) and the three w estimates were <1 for the large data set (Table 3). In the small data set, however, a w = 1 class was inferred (p = 22%) and one of the estimates for the large data set was close to one (w = 0.814, p = 26%). Therefore, it is still uncertain whether the existence of neutral sites is statistically supported, although there are clear indications that a number of sites have undergone relaxation of selective pressures during P3 evolution. Figure 2 illustrates that the N-terminal region of the protein is subjected to w values higher than the central region, which encloses many conserved sites. When we project the average posterior w values for each site inferred by the M3 (K = 4) model, which depicts the heterogeneity of selective pressures, on the primary structure of the CP S-domain, the N-terminal region of the sequence is also found to be composed of sites with higher w values, while the bulk of the sequence is under strong purifying selection (Figure 2). If average w’s in each of these regions are calculated using the average posterior w’s at each site (see Methods), the N-terminal region shows When the 3-D structure of Terradot et al. (2001) is considered, highly conserved sites (w = 0.052, M3 with K = 4 classes) are basically located within b-strands in the core of the structure and in regions facing the surface of the capsid (Figure 3A). The
This highly conserved structure of the surface of the capsid portrays potential interactions with cell receptors of the aphid vector. Mutational studies showed that sites under purifying selection on the CP surface are essential for the biology of the luteoviruses. For example, mutations in the acidic patch domain located in the surface loop of PLRV established the role of CP in virion assembly, systemic movement and aphid transmission (Lee et al., 2005). Mutation analyses of exposed sites of BWYV CP also recognized its function in RNA packaging, virus accumulation and aphid transmission. All amino acid sites deemed by Brault et al. (2003) and Lee et al. (2005) to be essential for well-functioning CP were found to be under negative selection in our analyses. Therefore, the purifying selection acting on the CP surface of Luteoviridae could be resultant of the constraints imposed by receptors of the aphid and the host plant, which limits structural changes. On the other hand, sites with NEB posterior probabilities >95% for w near neutrality (w = 1.169, M3 with K = 4 classes) are located at the region of the protein which faces the interior of the capsid (Figure 3A). Sites that make up the We have showed here that the heterogeneous selective forces acting on the CP of the Luteoviridae are in agreement with the studies that investigated the function of the protein. These indications lead us to find that evolutionary approaches, such as the analysis of differential selective pressures, can establish bridges between sequence data and biological function and can also be useful in identifying candidate sites for point mutation studies. ACKNOWLEDGMENTS M.W. Torres and R.L. Corrêa were supported by fellowships from CNPq (Brazilian research council). The authors wish to thank Laurent Terradot for providing the coordinates of the CP S-domain. This study was financially supported by research grants to Claudia A.M. Russo from CNPq and FAPERJ (Rio de Janeiro’s research foundation). REFERENCES Brault V, Bergdoll M, Mutterer J, Prasad V et al. (2003). Effects of point mutations in the major capsid protein of beet western yellows virus on capsid formation, virus accumulation, and aphid transmission. J. Virol. 77: 3247-3256. Choisy M, Woelk CH, Guegan JF and Robertson DL (2004). Comparative study of adaptive molecular evolution in different human immunodeficiency virus groups and subtypes. J. Virol. 78: 1962-1970. D’Arcy CJ and Mayo M (1997). Proposals for changes in luteovirus taxonomy and nomenclature. Arch. Virol. 142: 1285-1287. Dolja VV and Koonin EV (1991). Phylogeny of capsid proteins of small icosahedral RNA plant viruses. J. Gen. Virol. 72 (Pt. 7): 1481-1486. Domier LL, McCoppin NK, Larsen RC and D’Arcy CJ (2002). Nucleotide sequence shows that bean leafroll virus has a luteovirus-like genome organization. J. Gen. Virol. 83: 1791-1798. Frost SD, Gunthard HF, Wong JK, Havlir D et al. (2001). Evidence for positive selection driving the evolution of HIV-1 env under potent antiviral therapy. Virology 284: 250-258. Gray S and Gildow FE (2003). Luteovirus-aphid interactions. Annu. Rev. Phytopathol. 41: 539-566. Guyader S and Ducray DG (2002). Sequence analysis of Potato leafroll virus isolates reveals genetic stability, major evolutionary events and differential selection pressure between overlapping reading frame products. J. Gen. Virol. 83: 1799-1807. Habili N and Symons RH (1989). Evolutionary relationship between luteoviruses and other RNA plant viruses based on sequence motifs in their putative RNA polymerases and nucleic acid helicases. Nucleic Acids Res. 17: 9543-9555. Harisson BD (1999). Steps in the development of Luteovirology. In: The Luteoviridae (Smith H and Backer H, eds.). CABI Publishing, Wallingford, Oxon, England, pp. 1-14. Hasegawa M, Kishino H and Yano T (1985). Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22: 160-174. Lee L, Kaplan IB, Ripoll DR, Liang D et al. (2005). A surface loop of the potato leafroll virus coat protein is involved in virion assembly, systemic movement, and aphid transmission. J. Virol. 79: 1207-1214. Maia IG, Gonçalves MC, Arruda P and Vega J (2000). Molecular evidence that sugarcane yellow leaf virus (ScYLV) is a member of the Luteoviridae family. Arch. Virol. 145: 1009-1019. Mayo MA and Ziegler-Graff V (1996). Molecular biology of luteoviruses. Adv. Virus Res. 46: 413-460. McKee RK (1964). Virus infection in South America potatoes. Eur. Potato J. 7: 145-151. Mittal M, Tosh C, Hemadri D, Sanyal A et al. (2005). Phylogeny, genome evolution, and antigenic variability among endemic foot-and-mouth disease virus type A isolates from India. Arch. Virol. 150: 911-928. Moonan F, Molina J and Mirkov TE (2000). Sugarcane yellow leaf virus: an emerging virus that has evolved by recombination between luteoviral and poleroviral ancestors. Virology 269: 156-171. Oswald JW and Houston BR (1951). A new virus disease of cereals transmitted by aphids. Plant Dis. Rep. 35: 471-475. Peiffer ML, Gildow FE and Gray SM (1997). Two distinct mechanisms regulate luteovirus transmission efficiency and specificity at the aphid salivary gland. J. Gen. Virol. 78 (Pt. 3): 495-503. Posada D and Crandall KA (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14: 817-818. Rathjen JP, Karageorgos LE, Habili N, Waterhouse PM et al. (1994). Soybean dwarf luteovirus contains the third variant genome type in the luteovirus group. Virology 198: 671-679. Reutenauer A, Ziegler-Graff V, Lot H, Scheidecker D et al. (1993). Identification of beet western yellows luteovirus genes implicated in viral replication and particle morphogenesis. Virology 195: 692-699. Scott KP, Farmer MJ, Robinson DJ, Torrance L et al. (1996). Comparison of the coat protein of Groundnut rosette assistor virus with those of other luteoviruses. Ann. Appl. Biol. 128: 77-83. Shackelton LA, Parrish CR, Truyen U and Holmes EC (2005). High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. USA 102: 379-384. Smith GR, Borg Z, Lockhart BE, Braithwaite KS et al. (2000). Sugarcane yellow leaf virus: a novel member of the Luteoviridae that probably arose by inter-species recombination. J. Gen. Virol. 81: 1865-1869. Terradot L, Souchet M, Tran V and Giblot Ducray-Bourdin D (2001). Analysis of a three-dimensional structure of Potato leafroll virus coat protein obtained by homology modeling. Virology 286: 72-82. Thompson JD, Higgins DG and Gibson TJ (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673-4680. Wong WS, Yang Z, Goldman N and Nielsen R (2004). Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168: 1041-1051. Yang Z (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. CABIOS 13: 555-556. Yang Z (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15: 568-573. Yang Z, Nielsen R, Goldman N and Pedersen AM (2000). Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155: 431-449. |
|