![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
ABSTRACT. Genome annotation projects can produce incorrect results if they are based on obsolete data or inappropriate models. We have developed an automatic re-annotation system that uses agents to perform repetitive tasks and reports the results to the user. These tasks involve BLAST searches on biological databases (GenBank) and the use of detection tools (Genemark and Glimmer) to identify new open reading frames. Several agents execute these tools and combine their results to produce a list of open reading frames that is sent back to the user. Our goal was to reduce the manual work, executing most tasks automatically by computational tools. A prototype was implemented and validated using Mycoplasma pneumoniae and Haemophilus influenzae original annotated genomes. The results reported by the system identify most of new features present in the re-annotated versions of these genomes. Key words: Re-annotation, Bioinformatics, Agent-based systems INTRODUCTION Computers have an important role in DNA analysis. The use of computational tools reduces analysis time, through the processing of large amounts of data and through the integration of several approaches. Annotation by computational methods can execute repetitive and time-consuming tasks, speeding up the analysis of biological data. Current computational annotation methods are mainly based on comparative approaches. Computational tools, such as BLAST (Altschul et al., 1990) and its variants, search for homologous gene information stored in public biological databases. Positive hits are used in functional information inference by human experts to generate the annotation results. Other computational tools, such as Genemark (Borodovsky and McIninch, 1993) and GRAIL (Uberacher and Mural, 1991), use human knowledge models about DNA organization and signals for gene identification. Human knowledge about gene structure and DNA code organization is incomplete. Moreover, the information stored in biological databases used in annotation is periodically updated. Thus, genome annotation data can become obsolete and must be re-analyzed. These facts have stimulated many biologists to initiate re-annotation projects, in which the information acquired from the original annotation is revised and compared with new models and data. Despite the development of integrated annotation projects, there have been few efforts towards the automation of re-annotation processes. The existing projects have been based on the manual use of computational tools, such as BLAST. These tools compare new homologous genes and functional information with information available in biological databases. The results reported by these tools are manually analyzed and integrated by human experts. The use of an automatic re-annotation module would make the work easier and much faster. Specialized agents could automatically do many of the search and analysis tasks, leaving to the user the task of checking the results. We developed an integrated and automatic re-annotation system based on software agents. These agents use bioinformatics tools, searching biological databases and identifying new information. The user must register with a service, which informs, via e-mail, when a significant change in annotation has been found. RELATED WORK Re-annotation projects for individual species have been reported by a handful of groups. Most of them have used computational tools to identify new genes and to extend the information about annotated genes. The Haemophilus influenzae re-annotation project (Tatusov et al., 1996) used the BLASTX program to compare intergenic regions with entries in the Genbank database. A number of highly significant sequence similarities were encountered, indicating that these regions may contain additional genes. Given this new information, a revision was made of the set of proteins encoded by the H. influenzae genome. This re-annotation process combined sequence similarity searches with statistical analysis of the DNA sequences, using the Genemark program. This approach produced a new set of 1,703 putative protein-coding genes containing 23 new open reading frames (ORFs) and 107 modified ORFs. Moreover, 47 genes were eliminated because their existence could not be corroborated by any of the methods. In another recent project, the entire genome of Mycoplasma pneumoniae was re-annotated (Dandekar et al., 2000). The tasks involved in this project included comparisons with other genomes (in particular that of M. genitalium) and searches of biological databases, using tools such as PSI-BLAST (Altschul et al., 1997). The verification of results was made using software, such as HMMER (Durbin et al., 1998) and FASTA (Person and Lipman, 1998), as well as complementary tools and methods, such as domain analysis, phylogenetic analysis and analysis of context and clusters of orthologous genes. Experimental techniques, such as mass spectrometry and mRNA expression, were used as well. The re-annotated genome of the M. pneumoniae has 12 new proteins identified by the analysis of intergenic regions (two proteins identified by mass spectrometry, six hypothetical proteins and four with predicted functional features). Five other ORFs were eliminated because they contained pseudo-genes. The re-annotation process for the complete genome of Thermotoga maritima (Kyrpides et al., 2000) compared the 1,877 original ORFs with the corresponding new predictions. After discarding all cases where the two independent analyses agreed, cases of apparent disagreement and hypothetical proteins were analyzed in detail. The analysis used several computational tools, including:
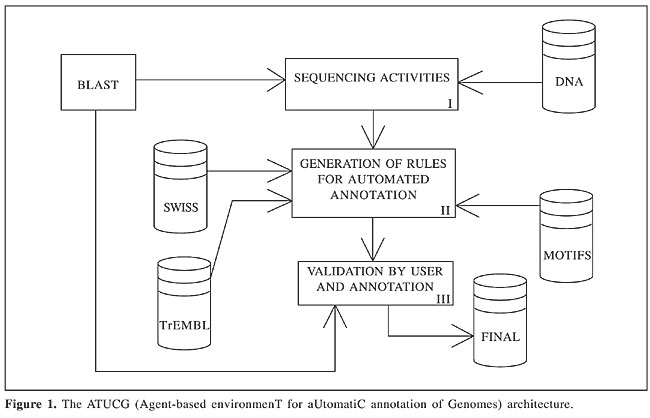
The conclusion was that 90% of the functional assignments agreed with the original ones. There were 193 new cases of conflicting annotations (10.3% of the entire genome), of which 164 were new function identifications and the remaining 29 cases were amendments to previously proposed functions. The total number of functional assignments increased from 1,014 (54%) to 1,178 (63%), which is a 16% increase. The ATUCG system The proposed re-annotation module has been included in an annotation system called ATUCG (Agent-based environmenT for aUtomatiC annotation of Genomes; Bazzan et al., 2003). The basic architecture of the ATUCG system is shown in Figure 1.
The system is composed of three layers. Layer I is responsible for building a non-redundant ORF list from the DNA sequence. This task is accomplished by the execution of several detection tools by individual agents. The results reported by all agents are merged and sent to the user for verification. The ORFs from layer I are analyzed in layer II. This layer executes a partial of the ORFs, assigning a key word list to each ORF. This functional annotation is obtained by classification rules that associate key words found in the Swiss-Prot database with motifs in the ORF sequence. The result is passed on to layer III, where the user can validate the annotation. The re-annotation module is inserted into layer I. Its function is to re-analyze the annotated ORFs stored in the database and periodically execute the re-annotation process. New annotations that are confirmed by the user are then stored in the system database. The re-annotation system The recent re-annotation projects used isolated computational tools, such as BLAST. Integration of the several tools used in the process was done manually by human experts. There is a lack of computational re-annotation tools that automatically execute the analysis programs and integrate their results. We are developing an automatic re-annotation system, based on multiagent-system technology. The system analyzes the annotated ORFs that are stored in a local database. The agents search for new annotations stored in public biological databases and they identify possible new genes. The analysis of annotated data is distributed and each agent executes a single task (Figure 2).
The proposed re-annotation approach uses three types of analysis:
The BLAST execution is configurable. The user can choose the BLAST variant (BLASTX, BLASTN, BLASTP, TBLASTX, or TBLASTN), the significance threshold and the sequence database used in the search. These agents make periodic analyses of annotated data. The results of the several agents are merged in a list reported to the user. This task is performed by the “Presentation agent”, which groups the BLAST hits by position relative to DNA sequence and annotation information. These groups contain hits with similar annotations and that are found in overlapping regions. Each group represents a possible re-annotation to an ORF, or a new ORF. The annotation comparisons are based on the functional domains associated with the regions of the hit. Overlapping hits that contain the same functional domains are placed in the same groups. The hit list in a group is ordered by significance, using the E value returned by BLAST. The user can choose to discard groups, manually edit the data, or accept the proposed re-annotation. Each agent follows the structure shown in Figure 3. The “Knowledge module” contains the information used by the agent, as well as the action results. These actions are executed by the “Action module”, which processes the execution requisitions. These requisitions are sent periodically by the system. These actions can be BLAST actions (which execute the NCBI BLAST tool), Intergenic processing actions or gene detection actions (Genemark, Glimmer). The “Communication module” receives the messages sent by the other agents and by the system. This module processes the incoming messages and sends them to the appropriate destinations. Action execution requisitions are sent to the “Action module”, while the data messages are sent to the “Knowledge module”. Agents exchange messages using FIPA ACL communication language.
RESULTS The results presented in this section were obtained from the analysis of original annotated versions of genome sequences retrieved from Genbank. The re-annotation process was executed over these sequences and the hits reported were manually compared with the re-annotated versions. The importance of the hits cannot be totally determined by the automatic analysis, and consequently some human validation is needed. We used the original M. pneumoniae ORFs reported in Himmelreich et al., 1996, shown in Table 1 (GI:6626256) and the ORFs in the H. influenzae genome (from bases 690806 to 702083 of the complete genome - GI:925682) for validation and evaluation of the results. The M. pneumoniae and H. influenzae genomes were recently re-annotated (Dandekar et al., 2000 and Tatusov et al., 1996). The re-annotated ORFs are available in Genbank (GI:13507739 and GI:1221355).
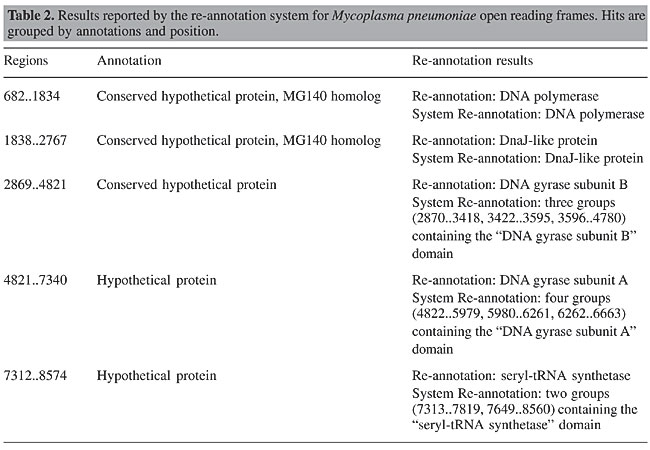
The original M. pneumoniae and H. influenzae ORFs were submitted to the BLAST and Intergenic agents. The BLAST tool was executed with an E value of 10-6 by both agents, using the Genbank non-redundant protein database. The results reported by these agents were analyzed by the Presentation agent. The hits reported by the automatic re-annotation system contained the same functional domains that are found in the re-annotated genome stored in Genbank. These functional domains were obtained from coding regions presented in the hit sequences. Table 2 shows re-annotations detected using the results reported by our system and the corresponding original re-annotated M. pneumoniae ORFs.
The results contain 1,339 hit groups. The ORFs detected using the groups proposed by the system and the original re-annotated ORFs have the same functional domains. The re-annotated M. pneumoniae genome has 689 ORFs. This difference between the number of hit groups and re-annotated ORFs is explained by the presence of redundant groups. For instance, the region between bases 2,869 and 4,821 contains three hit groups associated with it. An approach to avoid this problem is under development. The H. influenzae sequence was re-annotated and compared to a new version present in Genbank (GI:1573645). Table 3 shows the system re-annotation results. The analyzed sequence contains 11,286 bases. The only change found in the new version of the sequence is a new ORF between bases 8,716 and 9,252. This new ORF was detected by the re-annotation process. The region between 8,697 and 9,257 contains the “Topoisomerase DNA binding” functional domain, which agrees with the system re-annotation. The other regions that were analyzed did not contain hits indicating new annotations. This comparison was made manually from the hits reported by the system and from the Genbank data.
In the region between bases 823 and 1,099 one hit was reported that does not match any ORF in the H. influenzae re-annotation. New information reported by the system must be confirmed by human analysis. The relevance of the results (in the automatic re-annotation) is defined by the data reported by the computational tools that are used (BLAST, Genemark, Glimmer). Other types of result evaluation, such as chemical analysis, can only be performed by a human expert. CONCLUSION AND FUTURE WORK Automatic re-annotation is a useful process to detect changes in genome annotation. Human experts can analyze the results reported by several tools executed by software agents. These agents can run the tools in a distributed and integrated way, searching for new entries in sequence databases and using other tools on the original annotated sequences. Our re-annotation module uses multi-agent technology to re-analyze ORFs annotated using the ATUCG system. This module uses several agents running BLAST, Genemark and Glimmer tools on the annotated genome to identify new annotations. Intergenic regions are analyzed to identify new genes. The M. pneumoniae and H. influenzae genomes were used for evaluation purposes. The new ORFs reported by the system contain functional domains reported in the re-annotated versions. More tests will be executed in the future using Genemark and Glimmer agents on these DNA sequences and on other annotated genomes. We intend to use multiple alignment tools, such as HMMER, to extract motif and pattern information from BLAST hits in groups. The results will be compared to motif databases and will be used to add some functional information about the new sequences that are found. Conflicts can occur when the results reported by the agents are merged. Newly identified ORFs can overlap other annotated ORFs. Which are the correct ORFs? What are the criteria to be used to select the correct ones? Unfortunately most of the ORF selection work will need to be done by the user. However, this work can be facilitated by the pre-processing tasks performed by our system. We are studying an approach to treat the conflicts that can occur between the original annotated and the new re-annotated ORFs. ACKNOWLEDGMENTS Research supported by FAPERGS. L.V. Nascimento and A.L.C. Bazzan are partially supported by CNPq. REFERENCES Altschul, S., Gish, W., Miller, W., Myers, E. and Lipman, D. (1990). Basic local alignment search tool. J. Mol. Biol. 215: 403-410. Altschul, S., Madden, T., Shaffer, A., Zhang, Z., Miller, W. and Lipman, D. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. Bazzan, A., Duarte, R., Pitinga, A., Schroeder, L., Souto, F. and Da Silva, S. (2003). ATUCG - An Agent-Based Environment for Automatic Annotation of Genomes. IJCIS 12: 241-273. Benson, D., Karsch-Mizrachi, I., Lipman, D., Ostell, J. and Wheeler, D. (2004). GenBank: update. Nucleic Acids Res. 32: D23-D26. Borodovsky, M. and McIninch, J. (1993). GeneMark: parallel gene recognition for both DNA strands. Comput. Chem. 17: 123-133. Dandekar, T., Huynen, M., Regula, J., Ueberle, B., Zimmermann, C., Andrade, M., Doerks, T., Sánchez-Pulido, L., Snel, B., Suyama, M., Yuan, Y., Herrmann, R. and Bork, P. (2000). Re-annotating the Mycoplasma pneumoniae genome sequence: adding value, function and reading frames. Nucleic Acids Res. 28: 3278-3288. Durbin, R., Eddy, S., Krogh, A. and Mitchison, G. (1998). Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK. Himmelreich, R., Hilbert, H., Plagens, H., Pirkl, E., Li, B. and Herrmann, R. (1996). Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 24: 4420-4449. Kyrpides, N.C., Ouzounis, C.A., Iliopoulos, I., Vonstein, V. and Overbeek, R. (2000). Analysis of the Thermotoga maritima genome combining a variety of sequence similarity and genome context tools. Nucleic Acids Res. 28: 4573-4576. Person, W. and Lipman, D. (1998). Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85: 2444-2448. Tatusov, R.L., Mushegian, A.R., Bork, P., Brown, N.P., Hayes, W.S., Borodovsky, M., Rudd, K.E. and Koonin, E.V. (1996). Metabolism and evolution of Haemophilus influenzae deduced from a whole-genome comparison with Escherichia coli. Curr. Biol. 6: 279-291. Uberacher, E. and Mural, R. (1991). Locating protein-coding regions in human DNA sequences by a multiple sensor-neural network approach. Proc. Natl. Acad. Sci. USA 28: 11261-11265. |
|