![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
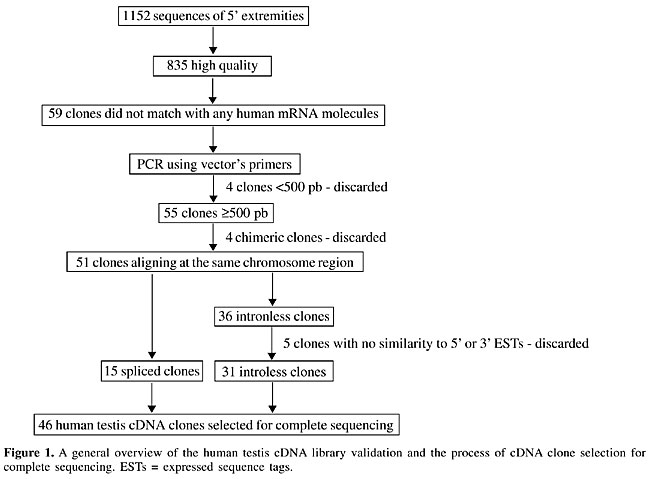
ABSTRACT. The correct identification of all human genes, and their derived transcripts, has not yet been achieved, and it remains one of the major aims of the worldwide genomics community. Computational programs suggest the existence of 30,000 to 40,000 human genes. However, definitive gene identification can only be achieved by experimental approaches. We used two distinct methodologies, one based on the alignment of mouse orthologous sequences to the human genome, and another based on the construction of a high-quality human testis cDNA library, in an attempt to identify new human transcripts within the human genome sequence. We generated 47 complete human transcript sequences, comprising 27 unannotated and 20 annotated sequences. Eight of these transcripts are variants of previously known genes. These transcripts were characterized according to size, number of exons, and chromosomal localization, and a search for protein domains was undertaken based on their putative open reading frames. In silico expression analysis suggests that some of these transcripts are expressed at low levels and in a restricted set of tissues. Key words: Novel human transcripts, Mouse orthologous sequence, Testis cDNA INTRODUCTION Due to the complexity of the human genome, the main objective of the Human Genome Project, the correct identification of all human genes and their derived transcripts, has yet to be achieved. The human genome is estimated to contain 30,000 to 40,000 genes (Lander et al., 2001; Venter et al., 2001). This number of genes is only slightly higher than what is found in much simpler organisms, such as Caenorhabditis elegans (The C. elegans Sequencing Consortium, 1998), suggesting that the greater complexity found in vertebrates might be due to a variety of mechanisms involving regulation of gene expression. Such mechanisms might include, for example, inhibition of gene expression by antisense transcripts (Shendure and Church, 2002; Yelin et al., 2003) and alternative splicing (Modrek and Lee, 2002). The coding sequences of the human genome, comprising only 3% of the entire sequence, are separated by large intergenic regions, and they are composed of short transcribed exons, normally interrupted by numerous, very long non-transcribed introns. Gene prediction programs have been used to recognize patterns and predict genes within the genome sequence. These programs are based not only on ab initio gene prediction, but they also make use of orthologous sequences, motif and domain structure, and the presence of polyadenylation sites and/or signals (Burge and Karlin, 1997; Rogic et al., 2001; Solovyev, 2001; Blanco et al., 2002). Despite the progress in this area, genomic structural complexity does not allow such programs to correctly predict all human genes without experimental confirmation. As a result, the correct identification of all human genes, and their derived transcripts, remains a real challenge. Many large-scale sequencing projects have contributed to the global identification of human genes and their variants by generating expressed sequence tags (ESTs) (Adams et al., 1991; Houlgatte et al., 1995) and open reading frame (ORF) ESTs (ORESTES) (Dias-Neto et al., 2000; de Souza et al., 2000; Camargo et al., 2001; Brentani et al., 2003). ESTs are partial and single-pass sequences derived from the 5’ or 3’ extremities of cDNA clones (Adams et al., 1991), whereas the ORESTES approach is biased towards the central portion of the coding regions of transcripts (Dias-Neto et al., 2000; Camargo et al., 2001). Nevertheless, full-length transcript sequences are crucial for final confirmation of gene structure. Considerable effort has been made in the generation of full-length transcript sequences (Strausberg et al., 1999; Wiemann et al., 2001; Kikuno et al., 2002; Nakajima et al., 2002; Strausberg et al., 2002) directly from high-quality cDNA libraries (Bonaldo et al., 1996; Carninci et al., 2000). The transcript finishing strategy, developed by Sogayar and collaborators (2004), utilized RT_PCR experiments to bridge gaps between EST clusters mapped to the human genome to achieve final confirmation of the structure of transcripts. Our involvement with this latter project motivated us to further explore the complete characterization of new human transcripts through integrative approaches involving experimental and in silico strategies. We used mouse transcript sequences and cDNA molecules derived from a human testis library to identify new human transcripts. We completed the sequence of 47 transcripts, including 27 that were first annotated by us in the human genome. MATERIAL AND METHODS Testis cDNA library generation and clone selection A unidirectional human testis cDNA library was constructed from polyA RNA using the SuperscriptTM Plasmid System GatewayTM Technology for double-strand cDNA synthesis and cloning. Cloned fragments were selected by size on Sepharose CL-2B (Pharmacia) columns (40 cm long, 1 mm ID) according to the protocol described by Vettore and collaborators (2001). Fractions containing cDNA molecules larger than 800 bp were ligated into pSPORT6 vectors (Invitrogen) at the SalI-NotI site and the resulting plasmids were transformed in DH10B cells (Invitrogen) by electroporation (BioRad). The transformants were spread on LB agar medium containing ampicilin (100 µg/ml), IPTG (100 mM) and X-Gal (20 mg/ml), and plasmid DNA was purified using the alkaline lysis method (Sambrook et al., 1989). In order to estimate the frequency of full-length clones, putative new transcripts, and the level of redundancy in the library, 5’ sequences from 192 clones were generated, resulting in 153 high-quality sequences (300 bp with Phred >20) suitable for further analysis. The 5’ sequences were aligned to the human genome using the BLAT search tool provided by the University of California, Santa Cruz (UCSC) (http://genome.ucsc.edu/cgi-bin/hgBlat, version Nov. 2002), and the annotation tracks corresponding to Known Genes, human mRNA and RefSeq genes were used for the comparison. The sequences that aligned with already identified full-length human mRNAs were used to estimate the frequency of full-length clones within the library. A sequence was considered a full-length clone when the 5’ end aligned with, or upstream of, the start codon site of a corresponding CDS-annotated mRNA molecule. The sequences that did not align with any full-length human mRNA were used to assess the frequency of putative new human transcripts. The CAP3 assembler program with default parameters (Huang and Madan, 1999) was used to join sequences with a high-identity level into contigs. The number of contigs and singletons obtained were divided by the total number of reads, and the redundancy level was assumed as 1 minus the value obtained in the previous calculation. cDNA evaluation and sequencing Inserts were amplified by PCR using the primers (SP6 Promoter primer - 5’ ATTTAGGTGACACTATA 3’ - and T7 Promoter primer - 5’ CCCTATAGTGAGTCGTATTA 3’) in a standard reaction containing 1X Taq polymerase buffer, 0.25 mM dNTP, 1.5 mM MgCl2, 1.0 mM each primers and 2 U Taq DNA polymerase (Invitrogen) in a final volume of 25 µl. Reactions were carried out at 94°C for 30 s, 55°C for 45 s and 72°C for 4 min for 35 cycles. An initial cycle of 94°C for 4 min and a final extension at 72° for 6 min were used. For generation of the complete sequence, two strategies were used: primer walking for clones smaller than 1,500 bp and shotgun libraries for clones larger than 1,500 bp. For the primer walking strategy, direct sequencing was undertaken, using internal primers designed approximately 100 bases from the end of the available high-quality sequence. For the shotgun strategy, the inserts were amplified by PCR and randomly fragmented by sonication. Fragments of 500 bp to 1,000 bp were isolated from agarose gels (1%) and cloned using the TOPO Shotgun Cloning Kit (Invitrogen). Identification of human transcripts using mouse cDNA sequences Mouse mRNA sequences available from UniGene were analyzed by the Laboratory of Computational Biology, Ludwig Institute for Cancer Research, São Paulo Branch. All mouse sequences were mapped onto the human genome sequence using Blast, and those that did not correspond to a full-length human mRNA were selected for further analysis. A total of 672 sequences of the list were manually checked, and those sequences that presented a functional annotation and size less than 4.0 kb were selected for evaluation by RT_PCR. RT_PCR amplification of human transcripts identified with mouse sequences Following selection of the mouse orthologs of interest, their sequences were aligned to the human genome sequence (http://genome.ucsc.edu/cgi-bin/hgBlat, version Nov. 2002), and RT_PCR primers were designed from conserved regions using Primer3 with default parameters (www-genome.wi.mit.edu/cgi-bin/primer/primer3.cgi). Since most sequences were larger than 500 bp, the construction of one or more pairs of primers was necessary to cover the entire transcript. Each pair of primers delimited a fragment of up to 1,000 bp, with 100 bp overlapping between pairs of primers to facilitate assembly of a consensus sequence. In order to determine from which tissue the cDNA should be used as the RT_PCR template, mouse mRNA sequences were aligned against dbEST (http://www.ncbi.nlm.nih.gov/BLAST/). PCR was undertaken using a mixture that contained 1X Taq polymerase buffer, 0.20 mM dNTP, 1.5 mM MgCl2, 1.0 mM primers, and 2 U Taq DNA polymerase (Invitrogen) in a final volume of 25 µl. Reactions were carried out with a basic cycle consisting of 94°C for 30 s for denaturating, primer annealing at calculated temperature for 45 s and extension for 1 min at 72°C for 35 cycles, together with an initial denaturating at 94°C for 4 min and a final extension at 72°C for 6 min. Modifications in the PCR conditions for candidates showing no specific amplification were attempted including the addition of betaine (1 M) (Henke et al., 1997), alteration of annealing temperature and adjustments of MgCl2 concentration. RNA extraction and cDNA synthesis Total RNA was prepared from cultured cells using the cesium chloride cushion technique (Chirgwin et al., 1979) and subsequently treated with 100 U DNAse I (FPLC-pure; Amersham). For cDNA synthesis, a reverse-transcriptase PCR was carried out, using 5 to 10 µg total RNA, oligo(dt)12-18, random primer and Superscript II (Invitrogen), according to the manufacturer’s instructions. Following the synthesis, cDNA molecules were treated with RNAse H. Genomic DNA contamination and cDNA quality were evaluated through PCR amplifications using primers annealing to intronic sequences flanking exon 12 of the hMLH-1 gene (forward: 5’TGGTGTCTCTAGTTCTGG3’ - reverse: 5’ CATTGTTGTAGTAGCTCTCG3’) and primers located at the 5’ extremity of the Notch 2 transcript (11,433 bp) (forward: 5’ACTGTGGCCAACCAAGTTCTC3’ - reverse: 5’CTCTCACAGGTGCTCCCTTC3’), respectively. Template preparation and DNA sequencing DNA templates were prepared for the testis cDNA clones in a 96-well plate, using the alkaline lysis method. Transcripts identified with mouse sequences were sequenced directly following purification of PCR products with the QIAquick PCR Purification Kit (QIAGEN). Sequencing reactions were carried out using ABI Prism BigDye Terminator v3.0 Cycle Sequencing Ready Reactions (Applied Biosystems) and an ABI Prism 3100 (Applied Biosystems). Sequencing analysis and database update Chromatograms were classified into testis cDNA sequences (TESTIS) or mouse-derived sequences (MO) and were processed with the PredPhrap package (including phred, phd2fasta and cross_match, in this order) with default parameters. Before the Phrap was called to assemble the data into one contig, the sequence reads were screened against the vector sequence. The contig sequences, and their respective chromatograms, were visually analyzed. Transcript characterization Consensus sequences were aligned against the May 2004 version of the human genome sequence available at UCSC Genome Browser (Kent et al., 2002), using the BLAT search tool (http://genome.ucsc.edu/cgi-bin/hgBlat). This allowed determination of overlap with known genes and gene predictions. A transcript was considered novel if its alignment coordinates did not match the coordinates of Known Genes, RefSeq Genes, MGC sequences, or human mRNA annotation tracks available through the BLAT search tool. A transcript was defined as a splicing variant if the alignments revealed intron retention and/or alternative exon usage when compared to sequences available in the databases cited above. The following tracks were used for comparison with prior gene prediction: Fgenesh++ (Solovyev, 2001), Geneid (Guigó et al., 1992) and Genscan (Burset and Guigó, 1996), available through the BLAT search tool in the July 2003 version. An exon was considered to be predicted if it aligned within the coordinates defined by any of the three gene prediction programs (not necessarily sharing borders) and a transcript was considered not predicted if none of the exons were predicted by any of the computer programs. The consensus sequences corresponding to newly identified transcripts were translated into amino acid sequences using TRANSLATE (http://us.expasy.org/tools/dna.html). The ORF sequences were searched against Pfam and Prosite databases by the Hits tool (http://hits.isb-sib.ch/) to determine putative protein domains. ORFs from the TESTIS cDNA clones were those with the longest amino acid sequences, containing at least 40 amino acids. In the case of the MO transcripts, ORFs were selected on the basis of their match with those in the mouse transcript. SAGE Virtual tags were assigned to the transcript sequences comprising the 10 bp downstream of the NlaIII site most proximal to the 3’ extremity. The extracted tags were then analyzed using serial analysis of gene expression (SAGE) genie (http://cgap.nci.nih.gov/SAGE) to generate a putative expression pattern for each transcript. RESULTS Library validation and selection of testis cDNA molecules The testis cDNA library contained 2 x 106 cDNA clones. Approximately 35% (35 of 99) of cDNA molecules tested in an initial sample were judged to be full-length sequences, based on alignment with CDS-annotated human transcripts available in the public databases. This percentage was not as high as that achieved using special protocols for obtaining full-length cDNA sequences (Carninci et al., 2000, 2001), but it was superior to what was obtained with commercially available, high-quality cDNA libraries evaluated by our group (data not shown). From an initial set of 153 sequences, we found 14 in which the 5’ extremity sequence did not match any known human transcript. This result indicated that up to 9% of our set might correspond to previously unidentified transcripts. Redundancy, as evaluated by CAP3 (Huang and Madan, 1999), was around 3.3%, with 145 singletons and three contigs. Based on these results the library was judged adequate to search for unknown transcripts. We then generated 1,152 5’ sequences, 835 of which were of high quality. Fifty-nine of these did not match known human transcripts, including 55 that were 500 bp or longer, based on PCR amplification. In order to detect chimeric clones, the sequences of both extremities were mapped to the Nov. 2002 version of the human genome sequence using BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat). Based on this, 4 clones were discarded, since their extremities did not align to the same genome region. Of the remaining 51 clones, 15 aligned discontinuously to the genomic sequence (revealing the presence of exons and introns), whereas 36 aligned continuously (indicating non-spliced structures). The presence of exons greatly increases the probability that the sequence is a bona fide transcript (Sorek and Safer, 2003). However, recent studies identified 3,500 single-exon human transcripts on the human genome sequence (approximately 10% of known genes) (Sakharkar and Kangueane, 2004), which appear to play an important role in transcript regulation and cell differentiation (Hickox et al., 2002; Sakharkar and Kangueane, 2004). We, thus, selected cDNA clones corresponding to putative single-exon transcripts where at least one corresponding EST was available in dbEST (http://www.ncbi.nlm.nih.gov/BLAST/). This was found to be the case for 31 sequences that mapped continuously to the genome. These and the 15 sequences corresponding to multi-exon transcripts were then submitted to complete sequencing (Figure 1).
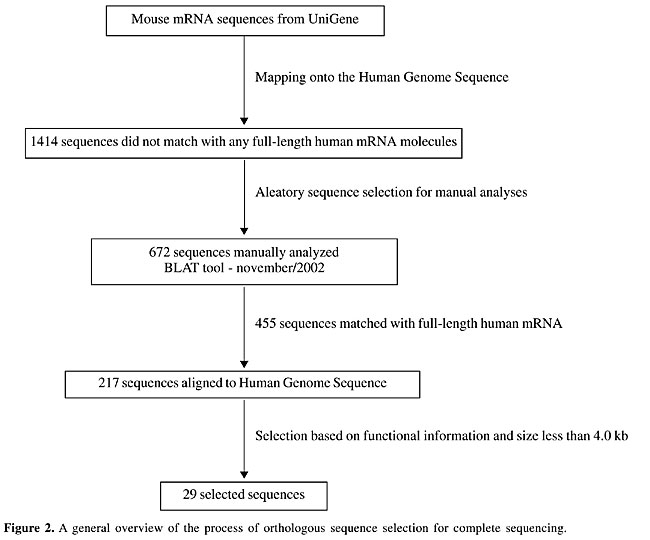
The use of mouse sequences to identify human transcripts One thousand four hundred and fourteen mouse sequences that had no match to any full-length human cDNA were considered. Manual inspection of 672 randomly selected sequences indicated that 217 had no alignment to any full-length human mRNA sequence in the public database. Several other criteria (See Methods) were then applied to reduce the dataset to a list of 29 regions in the human genome likely to contain a human transcript. An overview of the selection process is shown in Figure 2.
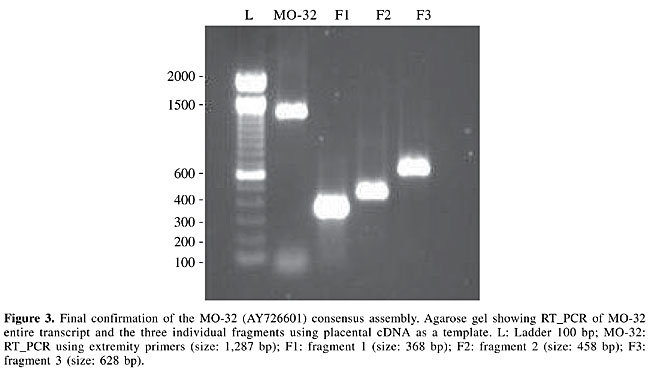
The source of ESTs matching those regions was used as a guide for tissue selection for PCR amplification. A pool of cDNA molecules from different tissues was used when no EST information was available. Among the 29 attempted amplifications, 28 generated at least one fragment of the expected size, indicating that as many as 96.5% of our candidates had a corresponding human ortholog. Consensus sequence assembly A total of 713 sequences were generated during the project, of which 305 were from TESTIS cDNA clones and 408 from the cDNA molecules identified with MO sequences; these were successfully assembled into 27 TESTIS cDNA molecules and 20 MO sequences. Fifteen TESTIS cDNA molecules and 6 MO sequences were abandoned once full-length human mRNA sequences were submitted to the GenBank by other groups. In the case of 4 TESTIS cDNA molecules and 3 MO sequences, it was impossible to generate a consensus sequence due to the presence of repetitive sequences (in the case of TESTIS cDNA molecules) or due to nonspecific amplification of fragments (in the case of MO sequences). Sequence discrepancies were manually corrected based on the genome sequence. Less than 0.9% of the nucleotides in the consensus assemblies required alteration. In the case of MO sequences, final confirmation came from RT_PCR, using primers annealing to the extremities of the consensus to demonstrate the existence of the complete transcript in the tissue (Figure 3). The 47 consensus sequences had an average size of 1,547 bp (1,721 bp for the TESTIS cDNA molecules and 1,312 bp for the MO sequences).
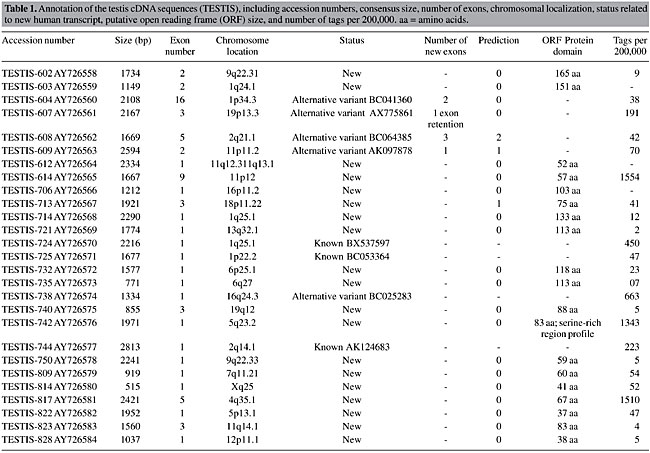
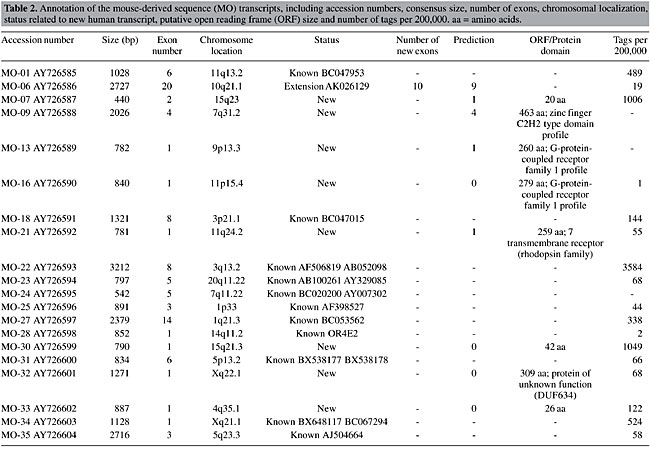
Analysis of the testis-derived transcripts Alignment of the 27 TESTIS cDNA transcripts to the human genome sequence (UCSC - May, 2004) revealed that 19 (70%) did not match full-length human mRNA sequences. Furthermore, only 5% of these presented a structure predicted by ab initio gene prediction programs (Fgenesh++, Geneid and Genscan), thus representing totally unknown transcribed regions in the human genome. Five TESTIS cDNA molecules comprised previously unknown splicing variants of known genes. Overall our data revealed 46 previously unidentified transcribed exons in the human genome, corresponding to 33,487 bp. The average size of the new exons was 727.9 bp. However, when single-exon transcripts were excluded the exon average size decreased to 438.0 bp (12 of 19 transcripts were single exon). Analysis of transcripts identified using mouse sequences A similar analysis for the 20 MO transcripts revealed that 8 did not match to full-length human mRNA sequences (40%) and one corresponded to a splicing variant of a known gene. Half of the novel transcripts had been predicted by ab initio gene prediction programs (Fgenesh++, Geneid and Genscan). Our data delimited 22 previously unknown exons in the human genome sequence comprising 9,060 bp. The average size of the new exons was 411.8 bp. When the six single-exon transcripts were excluded, the average exon size decreased to 231.8 bp. Transcript annotation Of the 27 full insert transcripts, 22 were found to contain an ORF of at least 40 amino acids, with the average ORF size being 129 amino acids. In 6 of these, the transcript contained an identifiable protein domain, such as a serine-rich region profile, a zinc finger C2H2 type domain and G-protein-coupled receptor family 1 profile. Amongst the MO transcripts, five of eight contained such profiles, while amongst the TESTIS transcripts the proportion was much lower, being 1 of 19. A complete listing of the characteristics of the TESTIS cDNAs is shown in Table 1 and another of the MO transcripts is shown in Table 2.
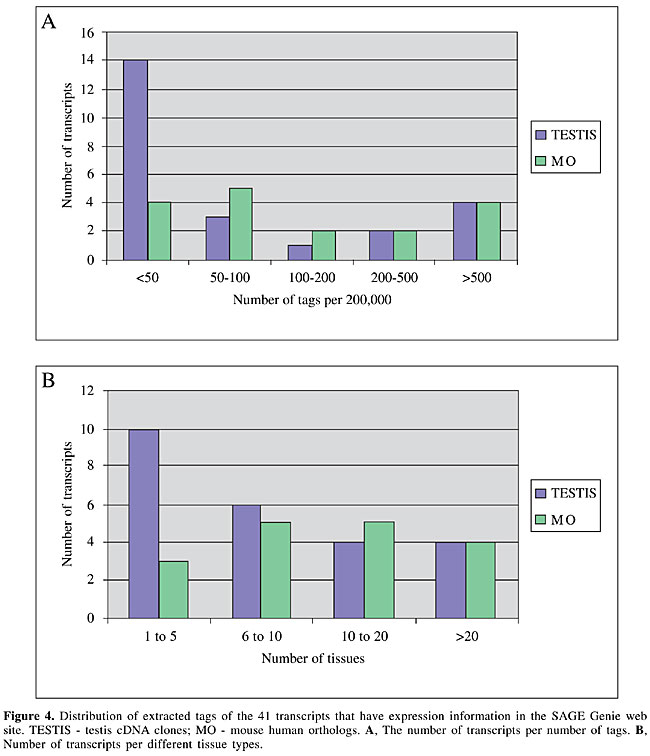
In silico analysis of transcript abundance In order to have a general view of the transcript expression profile from the human transcripts identified here, we performed an in silico analysis based on SAGE. The virtual tags corresponded to the 10 nucleotides immediately downstream of the 3’most NlaIII site in a given transcript. These virtual tags were then submitted to SAGE Genie (Boon et al., 2002) in order to establish transcript expression profiles (available at http://gdm.fmrp.usp.br/cgi-bin/tagmap/index.pl?template_file=view_sequence). We were able to obtain expression information for 41 of 47 sequences. Among these, 18 had less than 50 tags per 200,000, and were thus relatively lowly expressed (Figure 4A). Thirteen of the 41 transcripts were expressed in 5 or less tissues, as judged by the source of SAGE tags (Figure 4B). Amongst the TESTIS cDNA molecules analyzed, 14 of 24 had less than 50 tags per 200,000, whereas amongst the MO transcripts, only 4 of 17 presented this profile, and these were judged as rare messages. Similar results were found in tissue distribution analysis where 10 of 24 TESTIS cDNAs, and 3 of 17 MO transcripts were expressed in only 5 or less tissues.
DISCUSSION There is currently a worldwide effort to complete the catalogue of human genes and derived transcripts, involving several independent initiatives and distinct approaches. Full-length cDNA sequences are the gold standard for the definition of transcripts. Progress has been made in full-length sequence generation, using standard and full-length enriched cDNA library from many human tissues (Bonaldo et al., 1996; Carninci et al., 2000; Strausberg et al., 1999, 2002; Nakajima et al., 2002). A total of 28,256 UniGene clusters currently include at least one full-length cDNA sequence (UniGene Build #171 - http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene). To contribute to the definition of the human transcript catalogue, we have used two alternative strategies: the construction of a unidirectional human testis cDNA library and the alignment of mouse sequences to the human genome. Testis is a highly specialized tissue that expresses a large number of transcript species, which makes it suitable as a potential source for unidentified transcripts (Warrington et al., 2000; Yao et al., 2004). The strategy using mouse sequences is powerful, due to the high degree of conservation between human and mouse observed both in the coding sequence (Makalowski et al., 1996) and in 3’ and 5’ UTR (Makalowski et al., 1996; Shabalina et al., 2004). We identified and completely sequenced 47 previously unknown human transcripts, of which 27 had still not been annotated in the May 2004 version of the UCSC genome browser. The use of a cDNA testis library was found to be more effective (19 novel transcripts) than the use of mouse mRNA sequences (8 novel transcripts) for the identification of unknown human transcripts. Intronless transcripts have increasingly been perceived as playing an important role in the regulation of transcription (Hickox et al., 2002; Sakharkar and Kangueane, 2004), and they represent a significant proportion of the human gene catalogue (Sakharkar et al., 2002; Sakharkar and Kangueane, 2004). A significant proportion of our candidates were intronless transcripts (24 of 47). Many of them had still not been reported by others by the end of the project (18 of 24 intronless transcripts and 9 of 23 transcripts with multiple exons). The high number of novel intronless transcripts identified in this project may be due to the fact that many groups have been using strategies for transcript identification that exclude intronless sequences (Sogayar et al, 2004). Our analysis of the novel transcripts revealed that 22 of 27 were not identified by any of the commonly used ab initio gene prediction programs. Amongst the TESTIS transcripts, 18 of 19 were not predicted, while in the case of MO transcripts 4 of 8 were not predicted. This leads us to suggest that most of the unidentified transcripts have atypical structures that are difficult to identify using ab initio computational prediction programs. Amongst the novel human transcripts, only 9 of 27 had an ORF of more than 100 amino acids, precluding extensive analysis of predicted protein structure and function. Short ORF transcripts may be more difficult to predict by computational programs, and consequently their experimental characterization might have been delayed. Moreover, only 6 transcripts exhibited recognizable protein domains, 5 of which were MO transcripts. Whether the transcripts containing short ORFs without the presence of a known protein domain produce a functional protein remains to be determined. Recent technological advances in large-scale gene expression analysis have been made, including SAGE (Velculescu et al., 1995). Currently there are around 15 million tags available in the SAGE Genie database. These tags can be associated with a human transcript, providing a global expression portrait of the human genome, and the use of bioinformatics tools allows a general view of individual transcript distribution. The TESTIS cDNA molecules had a higher frequency of transcripts expressed at a low level and a restricted number of tissue types, when compared to MO transcripts. This result is supported by the higher proportion of novel human transcripts identified by the testis library approach, since this expression pattern could have made their previous identification more difficult. We conclude that there are still unidentified human transcripts, many of which might be found using the testis as a tissue source. Surprisingly, we also found that even genes fully annotated in the mouse genome remained cryptic in the human genome, although many of these transcripts either may not encode proteins or they could produce rather short polypeptides. Nevertheless, continued efforts at human gene identification would appear to be worthwhile. ACKNOWLEDGMENTS We thank Dr. Ricardo R. Brentani (Director of the Ludwig Institute-São Paulo Branch and of the A.C. Camargo Hospital) for valuable support. We also thank Anna Christina de Matos Salim, Elisangela Monteiro, Jane Kaiano, Dr. Maria Rita Passos Bueno, Guilherme M. Orabona, and Elisson Campos Osório for technical and computational assistance and Natanja Slager for critical reading and important comments on this manuscript. Research equally supported by the Ludwig Institute for Cancer Research and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). REFERENCES Adams, M.D., Kelley, J.M., Gocayne, J.D., Dubnick, M., Polymeropoulos, M.H., Xiao, H., Merril, C.R., Wu, A., Olde, B. and Moreno, R.F. (1991). Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 252: 1651-1656. Blanco, E., Parra, G. and Guigó, R. (2002). Finding genes. In: Current Protocols in Bioinformatics (Baxevanis, A., ed.). John Wiley & Sons Ltd., New York (in press). Bonaldo, M.F., Lennon, G. and Soares, M.B. (1996). Normalization and subtraction: two approaches to facilitate gene discovery. Genome Res. 6: 791-806. Boon, K., Osorio, E.C., Greenhut, S.F., Schaefer, C.F., Shoemaker, J., Polyak, K., Morin, P.J., Buetow, K.H., Strausberg, R.L., De Souza, S.J. and Riggins, G.J. (2002). An anatomy of normal and malignant gene expression. Proc. Natl. Acad. Sci. USA 99: 11287-11292. Brentani, H., Caballero, O.L., Camargo, A.A., da Silva, A.M., da Silva Jr., W.A., Dias Neto, E., Grivet, M., Gruber, A., Guimarães, P.E., Hide, W., Iseli, C., Jongeneel, C.V., Kelso, J., Nagai, M.A., Ojopi, E.P., Osório, E.C., Reis, E.M., Riggins, G.J., Simpson, A.J., de Souza, S., Stevenson, B.J., Strausberg, R.L., Tajara, E.H., Verjovski-Almeida, S., Acencio, M.L., Bengtson, M.H., Bettoni, F., Bodmer, W.F., Briones, M.R., Camargo, L.P., Cavenee, W., Cerutti, J.M., Coelho Andrade, L.E., Costa dos Santos, P.C., Ramos Costa, M.C., da Silva, I.T., Estecio, M.R., Sa Ferreira, K., Furnari, F.B., Faria Jr., M., Galante, P.A., Guimarães, G.S., Holanda, A.J., Kimura, E.T., Leerkes, M.R., Lu, X., Maciel, R.M., Martins, E.A., Massirer, K.B., Melo, A.S., Mestriner, C.A., Miracca, E.C., Miranda, L.L., Nóbrega, F.G., Oliveira, P.S., Paquola, A.C., Pandolfi, J.R., Campos Pardini, M.I., Passetti, F., Quackenbush, J., Schnabel, B., Sogayar, M.C., Souza, J.E., Valentini, S.R., Zaiats, A.C., Amaral, E.J., Arnaldi, L.A., de Araujo, A.G., de Bessa, S.A., Bicknell, D.C., Ribeiro de Camaro, M.E., Carraro, D.M., Carrer, H., Carvalho, A.F., Colin, C., Costa, F., Curcio, C., Guerreiro da Silva, I.D., Pereira da Silva, N., Dellamano, M., El-Dorry, H., Espreafico, E.M., Scattone Ferreira, A.J., Ayres Ferreira, C., Fortes, M.A., Gama, A.H., Giannella-Neto, D., Giannella, M.L., Giorgi, R.R., Goldman, G.H., Goldman, M.H., Hackel, C., Ho, P.L., Kimura, E.M., Kowalski, L.P., Krieger, J.E., Leite, L.C., Lopes, A., Luna, A.M., Mackay, A., Mari, S.K., Marques, A.A., Martins, W.K., Montagnini, A., Mourao Neto, M., Nascimento, A.L., Neville, A.M., Nobrega, M.P., O’Hare, M.J., Otsuka, A.Y., Ruas de Melo, A.I., Paco-Larson, M.L., Guimaraes Pereira, G., Pereira da Silva, N., Pesquero, J.B., Pessoa, J.G., Rahal, P., Rainho, C.A., Rodrigues, V., Rogatto, S.R., Romano, C.M., Romeiro, J.G., Rossi, B.M., Rusticci, M., Guerra de Sa, R., Sant’ Anna, S.C., Sarmazo, M.L., Silva, T.C., Soares, F.A., Sonati, M. de F., de Freitas Sousa, J., Queiroz, D., Valente, V., Vettore, A.L., Villanova, F.E., Zago, M.A., Zalcberg, H. and the Human Cancer Genome Project/Cancer Genome Anatomy Project Annotation Consortium; Human Cancer Genome Project Sequencing Consortium (2003). The generation and utilization of a cancer-oriented representation of the human transcriptome by using expressed sequence tags. Proc. Natl. Acad. Sci. USA 100: 13418-13423. Burge, C. and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. Burset, M. and Guigó, R. (1996). Evaluation of gene structure prediction programs. Genomics 34: 353-367. Camargo, A.A., Samaia, H.P., Dias-Neto, E., Simao, D.F., Migotto, I.A., Briones, M.R., Costa, F.F., Nagai, M.A., Verjovski-Almeida, S., Zago, M.A., Andrade, L.E., Carrer, H., El-Dorry, H.F., Espreafico, E.M., Habr-Gama, A., Giannella-Neto, D., Goldman, G.H., Gruber, A., Hackel, C., Kimura, E.T., Maciel, R.M., Marie, S.K., Martins, E.A., Nobrega, M.P., Paco-Larson, M.L., Pardini, M.I., Pereira, G.G., Pesquero, J.B., Rodrigues, V., Rogatto, S.R., da Silva, I.D., Sogayar, M.C., Sonati, M.F., Tajara, E.H., Valentini, S.R., Alberto, F.L., Amaral, M.E., Aneas, I., Arnaldi, L.A., de Assis, A.M., Bengtson, M.H., Bergamo, N.A., Bombonato, V., de Camargo, M.E., Canevari, R.A., Carraro, D.M., Cerutti, J.M., Correa, M.L., Correa, R.F., Costa, M.C., Curcio, C., Hokama, P.O., Ferreira, A.J., Furuzawa, G.K., Gushiken, T., Ho, P.L., Kimura, E., Krieger, J.E., Leite, L.C., Majumder, P., Marins, M., Marques, E.R., Melo, A.S., Barbosa de Melo, M., Mestriner, C.A., Miracca, E.C., Miranda, D.C., Nascimento, A.L., Nobrega, F.G., Ojopi, E.P., Pandolfi, J.R., Pessoa, L.G., Prevedel, A.C., Rahal, P., Rainho, C.A., Reis, E.M., Ribeiro, M.L., da Ros, N., de Sa, R.G., Sales, M.M., Sant’anna, S.C., dos Santos, M.L., da Silva, A.M., da Silva, N.P., Silva Jr., W.A., da Silveira, R.A., Sousa, J.F., Stecconi, D., Tsukumo, F., Valente, V., Soares, F., Moreira, E.S., Nunes, D.N., Correa, R.G., Zalcberg, H., Carvalho, A.F., Reis, L.F., Brentani, R.R., Simpson, A.J., de Souza, S.J. and Melo, M.B. (2001). The contribution of 700,000 ORF sequence tags to the definition of the human transcriptome. Proc. Natl. Acad. Sci. USA 98: 12103-12108. Carninci, P., Shibata, Y., Hayatsu, N., Sugahara, Y., Shibata, K., Itoh, M., Konno, H., Okazaki, Y., Muramatsu, M. and Hayashizaki, Y. (2000). Normalization and subtraction of cap-trapper-selected cDNA molecules to prepare full-length cDNA libraries for rapid discovery of new genes. Genome Res. 10: 1617-1630. Carninci, P., Shibata, Y., Hayatsu, N., Itoh, M., Shiraki, T., Hirozane, T., Watahiki, A., Shibata, K., Konno, H., Muramatsu, M. and Hayashizaki, Y. (2001). Balanced-size and long-size cloning of full-length, cap-trapped cDNAs into vectors of the novel lambda-FLC family allows enhanced gene discovery rate and functional analysis. Genomics 77: 79-90. Chirgwin, J.M., Przybyla, A.E., McDonald, R.J. and Rutter W.J. (1979). Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18: 5294-5299. de Souza, S.J., Camargo, A.A., Briones, M.R., Costa, F.F., Nagai, M.A., Verjovski-Almeida, S., Zago, M.A., Andrade, L.E., Carrer, H., El-Dorry, H.F., Espreafico, E.M., Habr-Gama, A., Giannella-Neto, D., Goldman, G.H., Gruber, A., Hackel, C., Kimura, E.T., Maciel, R.M., Marie, S.K., Martins, E.A., Nóbrega, M.P., Paco-Larson, M.L., Pardini, M.I., Pereira, G.G., Pesquero, J.B., Rodrigues, V., Rogatto, S.R., da Silva, I.D., Sogayar, M.C., de Fatima Sonati, M., Tajara, E.H., Valentini, S.R., Acencio, M., Alberto, F.L., Amaral, M.E., Aneas, I., Bengtson, M.H., Carraro, D.M., Carvalho, A.F., Carvalho, L.H., Cerutti, J.M., Correa, M.L., Costa, M.C., Curcio, C., Gushiken, T., Ho, P.L., Kimura, E., Leite, L.C., Maia, G., Majumder, P., Marins, M., Matsukuma, A., Melo, A.S., Mestriner, C.A., Miracca, E.C., Miranda, D.C., Nascimento, A.N., Nobrega, F.G., Ojopi, E.P., Pandolfi, J.R., Pessoa, L.G., Rahal, P., Rainho, C.A., da Ros, N., de Sa, R.G., Sales, M.M., da Silva, N.P., Silva, T.C., da Silva Jr., W., Simao, D.F., Sousa, J.F., Stecconi, D., Tsukumo, F., Valente, V., Zalcbeg, H., Brentani, R.R., Reis, F.L., Dias-Neto, E. and Simpson, A.J. (2000). Identification of human chromosome 22 transcribed sequences with ORF expressed sequence tags. Proc. Natl. Acad. Sci. USA 97: 12690-12693. Dias-Neto, E., Correa, R.G., Verjovski-Almeida, S., Briones, M.R., Nagai, M.A., da Silva Jr., W., Zago, M.A., Bordin, S., Costa, F.F., Goldman, G.H., Carvalho, A.F., Matsukuma, A., Baia, G.S., Simpson, D.H., Brunstein, A., de Oliveira, P.S., Bucher, P., Jongeneel, C.V., O’Hare, M.J., Soares, F., Brentani, R.R., Reis, L.F., de Souza, S.J. and Simpson, A.J. (2000). Shotgun sequencing of the human transcriptome with ORF expressed sequence tags. Proc. Natl. Acad. Sci. USA 97: 3491-3496. Guigó, R., Knudsen, S., Drake, N. and Smith, T. (1992). Prediction of gene structure. J. Mol. Biol. 226: 141-157. Henke, W., Herdel, K., Jung, K., Schoor, D. and Lorning, S.A. (1997). Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res. 25: 3957-3958. Hickox, D.M., Gibbs, G., Morrison, J.R., Sebire, K., Edgar, K., Keah, H.H., Alter, K., Loveland, K.L., Hearn, M.T.W., de Kretser, D.M. and O’Bryan, M.K. (2002). Identification of a novel testis-specific member of the phosphatidylethanolamine binding protein family, pebp-2. Biol. Reprod. 67: 917-927. Houlgatte, R., Mariage-Samson, R., Duprat, S., Tessier, A., Bentolila, S., Lamy, B. and Auffray, C. (1995). The Genexpress Index: a resource for gene discovery and the genic map of the human genome. Genome Res. 5: 272-304. Huang, X. and Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9: 868-877. Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M., Pringle, T.H., Zahler, A.M. and Haussler, D. (2002). The Human Genome Browser at UCSC. Genome Res. 12: 996-1006. Kikuno, R., Nagase, T., Waki, M. and Ohara, O. (2002). HUGE: a database for human large proteins identified in the Kazusa cDNA sequencing project. Nucleic Acids Res. 30: 166-168. Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., McKernan, K., Meldrim, J., Mesirov, J.P., Miranda, C., Morris, W., Naylor, J., Raymond, C., Rosetti, M., Santos, R., Sheridan, A., Sougnez, C., Stange-Thomann, N., Stojanovic, N., Subramanian, A., Wyman, D., Rogers, J., Sulston, J., Ainscough, R., Beck, S., Bentley, D., Burton, J., Clee, C., Carter, N., Coulson, A., Deadman, R., Deloukas, P., Dunham, A., Dunham, I., Durbin, R., French, L., Grafham, D., Gregory, S., Hubbard, T., Humphray, S., Hunt, A., Jones, M., Lloyd, C., McMurray, A., Matthews, L., Mercer, S., Milne, S., Mullikin, J.C., Mungall, A., Plumb, R., Ross, M., Shownkeen, R., Sims, S., Waterston, R.H., Wilson, R.K., Hillier, L.W., McPherson, J.D., Marra, M.A., Mardis, E.R., Fulton, L.A., Chinwalla, A.T., Pepin, K.H., Gish, W.R., Chissoe, S.L., Wendl, M.C., Delehaunty, K.D., Miner, T.L., Delehaunty, A., Kramer, J.B., Cook, L.L., Fulton, R.S., Johnson, D.L., Minx, P.J., Clifton, S.W., Hawkins, T., Branscomb, E., Predki, P., Richardson, P., Wenning, S., Slezak, T., Doggett, N., Cheng, J.F., Olsen, A., Lucas, S., Elkin, C., Uberbacher, E., Frazier, M., Gibbs, R.A., Muzny, D.M., Scherer, S.E., Bouck, J.B., Sodergren, E.J., Worley, K.C., Rives, C.M., Gorrell, J.H., Metzker, M.L., Naylor, S.L., Kucherlapati, R.S., Nelson, D.L., Weinstock, G.M., Sakaki, Y., Fujiyama, A., Hattori, M., Yada, T., Toyoda, A., Itoh, T., Kawagoe, C., Watanabe, H., Totoki, Y., Taylor, T., Weissenbach, J., Heilig, R., Saurin, W., Artiguenave, F., Brottier, P., Bruls, T., Pelletier, E., Robert, C., Wincker, P., Smith, D.R., Doucette-Stamm, L., Rubenfield, M., Weinstock, K., Lee, H.M., Dubois, J., Rosenthal, A., Platzer, M., Nyakatura, G., Taudien, S., Rump, A., Yang, H., Yu, J., Wang, J., Huang, G., Gu, J., Hood, L., Rowen, L., Madan, A., Qin, S., Davis, R.W., Federspiel, N.A., Abola, A.P., Proctor, M.J., Myers, R.M., Schmutz, J., Dickson, M., Grimwood, J., Cox, D.R., Olson, M.V., Kaul, R., Raymond, C., Shimizu, N., Kawasaki, K., Minoshima, S., Evans, G.A., Athanasiou, M., Schultz, R., Roe, B.A., Chen, F., Pan, H., Ramser, J., Lehrach, H., Reinhardt, R., McCombie, W.R., de la Bastide, M., Dedhia, N., Blocker, H., Hornischer, K., Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J.A., Bateman, A., Batzoglou, S., Birney, E., Bork, P., Brown, D.G., Burge, C.B., Cerutti, L., Chen, H.C., Church, D., Clamp, M., Copley, R.R., Doerks, T., Eddy, S.R., Eichler, E.E., Furey, T.S., Galagan, J., Gilbert, J.G., Harmon, C., Hayashizaki, Y., Haussler, D., Hermjakob, H., Hokamp, K., Jang, W., Johnson, L.S., Jones, T.A., Kasif, S., Kaspryzk, A., Kennedy, S., Kent, W.J., Kitts, P., Koonin, E.V., Korf, I., Kulp, D., Lancet, D., Lowe, T.M., McLysaght, A., Mikkelsen, T., Moran, J.V., Mulder, N., Pollara, V.J., Ponting, C.P., Schuler, G., Schultz, J., Slater, G., Smit, A.F., Stupka, E., Szustakowski, J., Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler, R., Williams, A., Wolf, Y.I., Wolfe, K.H., Yang, S.P., Yeh, R.F., Collins, F., Guyer, M.S., Peterson, J., Felsenfeld, A., Wetterstrand, K.A., Patrinos, A., Morgan, M.J., Szustakowki, J., de Jong, P., Catanese, J.J., Osoegawa, K., Shizuya, H., Choi, S., Chen, Y.J. and the International Human Genome Sequencing Consortium (2001). Initial sequencing and analysis of the human genome. Nature 409: 860-921. Liang, F., Holt, I., Pertea, G., Karamycheva, S., Salzberg, S.L. and Quackenbush, J. (2000). Gene index analysis of the human genome estimates approximately 120,000 genes. Nature 25: 239-240. Makalowski, W., Zhang, J. and Boguski, M.S. (1996). Comparative analysis of 1196 orthologous mouse and human full-length mRNA and protein sequences. Genome Res. 6: 846-857. Modrek, B. and Lee, C. (2002) A genomic view of alternative splicing. Nat. Genet. 30: 13-19. Nakajima, D., Okazaki, N., Yamakawa, H., Kikuno, R., Ohara, O. and Nagase, T. (2002). Construction of expression-ready cDNA clones for KIAA genes: Manual curation of 330 KIAA cDNA clones. DNA Res. 9: 99-106. Rogic, S., Mackworth, A.K. and Ouellette, F.B. (2001). Evaluation of gene-finding programs on mammalian sequences. Genome Res. 11: 817-832. Sakharkar, M.K. and Kangueane, P. (2004). Genome SEGE: A database for ‘intronless’ genes in eukaryotic genomes. BMC Bioinformatics 5: 67. Sakharkar, M.K., Kangueane, P., Petrov, D.A., Kolaskar, A.S. and Subbiah, S. (2002). SEGE: A database on ‘intron less/single exonic’ genes from eukaryotes. Bioinformatics 18: 1266-1267. Sambrook, J., Fritsch, E. and Maniatis, T. (1989). Molecular Cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA. Shabalina, S.A., Ogurtsov, A.Y., Rogozin, I.B., Koonin, E.V. and Lipman, D.J. (2004). Comparative analysis of orthologous eukaryotic mRNAs: potential hidden functional signals. Nucleic Acids Res. 32: 1774-1782. Shendure, J. and Church, G.M. (2002). Computational discovery of sense-antisense transcription in the human and mouse genomes. Genome Biol. 3: 1-14. Sogayar, M.C., Camargo, A.A., Bettoni, F., Carraro, D.M., Pires, L.C., Parmigiani, R.B., Ferreira, E.N., de Sa Moreira, E., do Rosario D de O Latorre, M., Simpson, A.J., Cruz, L.O., Degaki, T.L., Festa, F., Massirer, K.B., Sogayar, M.C., Filho, F.C., Camargo, L.P., Cunha, M.A., De Souza, S.J., Faria Jr., M., Giuliatti, S., Kopp, L., de Oliveira, P.S., Paiva, P.B., Pereira, A.A., Pinheiro, D.G., Puga, R.D., S de Souza, J.E., Albuquerque, D.M., Andrade, L.E., Baia, G.S., Briones, M.R., Cavaleiro-Luna, A.M., Cerutti, J.M., Costa, F.F., Costanzi-Strauss, E., Espreafico, E.M., Ferrasi, A.C., Ferro, E.S., Fortes, M.A., Furchi, J.R., Giannella-Neto, D., Goldman, G.H., Goldman, M.H., Gruber, A., Guimaraes, G.S., Hackel, C., Henrique-Silva, F., Kimura, E.T., Leoni, S.G., Macedo, C., Malnic, B., Manzini, B.C.V., Marie, S.K., Martinez-Rossi, N.M., Menossi, M., Miracca, E.C., Nagai, M.A., Nobrega, F.G., Nobrega, M.P., Oba-Shinjo, S.M., Oliveira, M.K., Orabona, G.M., Otsuka, A.Y., Paco-Larson, M.L., Paixao, B.M., Pandolfi, J.R., Pardini, M.I., Passos Bueno, M.R., Passos, G.A., Pesquero, J.B., Pessoa, J.G., Rahal, P., Rainho, C.A., Reis, C.P., Ricca, T.I., Rodrigues, V., Rogatto, S.R., Romano, C.M., Romeiro, J.G., Rossi, A., Sa, R.G., Sales, M.M., Sant’Anna, S.C., Santarosa, P.L., Segato, F., Silva Jr., W.A., Silva, I.D., Silva, N.P., Soares-Costa, A., Sonati, M.F., Strauss, B.E., Tajara, E.H., Valentini, S.R., Villanova, F.E., Ward, L.S., Zanette, D.L. and the Ludwig-FAPESP Transcript Finishing Initiative (2004). A transcript finishing initiative for closing gaps in the human transcriptome. Genome Res. 14: 1413-1423. Solovyev, V. (2001). Statistical approaches in eukaryotic gene prediction. In: Handbook of Statistical Genetics (Balding, D.J., Bishop, M. and Cannings, C., eds.). John Wiley & Sons Ltd., UK, pp. 83-127. Sorek, R. and Safer, H.M. (2003). A novel algorithm for computational identification of contaminated EST libraries. Nucleic Acids Res. 31: 1067-1074. Strausberg, R.L., Feingold, E.A., Klausner, R.D. and Collins, F.S. (1999). The mammalian gene collection. Science 286: 455-457. Strausberg, R.L., Feingold, E.A., Grouse, L.H., Derge, J.G., Klausner, R.D., Collins, F.S., Wagner, L., Shenmen, C.M., Schuler, G.D., Altschul, S.F., Zeeberg, B., Buetow, K.H., Schaefer, C.F., Bhat, N.K., Hopkins, R.F., Jordan, H., Moore, T., Max, S.I., Wang, J., Hsieh, F., Diatchenko, L., Marusina, K., Farmer, A.A., Rubin, G.M., Hong, L., Stapleton, M., Soares, M.B., Bonaldo, M.F., Casavant, T.L., Scheetz, T.E., Brownstein, M.J., Usdin, T.B., Toshiyuki, S., Carninci, P., Prange, C., Raha, S.S., Loquellano, N.A., Peters, G.J., Abramson, R.D., Mullahy, S.J., Bosak, S.A., McEwan, P.J., McKernan, K.J., Malek, J.A., Gunaratne, P.H., Richards, S., Worley, K.C., Hale, S., Garcia, A.M., Gay, L.J., Hulyk, S.W., Villalon, D.K., Muzny, D.M., Sodergren, E.J., Lu, X., Gibbs, R.A., Fahey, J., Helton, E., Ketteman, M., Madan, A., Rodrigues, S., Sanchez, A., Whiting, M., Madan, A., Young, A.C., Shevchenko, Y., Bouffard, G.G., Blakesley, R.W., Touchman, J.W., Green, E.D., Dickson, M.C., Rodriguez, A.C., Grimwood, J., Schmutz, J., Myers, R.M., Butterfield, Y.S., Krzywinski, M.I., Skalska, U., Smailus, D.E., Schnerch, A., Schein, J.E., Jones, S.J., Marra M.A. and the Mammalian Gene Collection Program Team (2002). Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA 99: 16899-16903. The C. elegans Sequencing Consortium (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012-2018. Velculescu, V.E., Zhang, L., Vogelstein, B. and Kinzler, K.W. (1995). Serial analysis of gene expression. Science 270: 484-487. Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., Gocayne, J.D., Amanatides, P., Ballew, R.M., Huson, D.H., Wortman, J.R., Zhang, Q., Kodira, C.D., Zheng, X.H., Chen, L., Skupski, M., Subramanian, G., Thomas, P.D., Zhang, J., Gabor Miklos, G.L., Nelson, C., Broder, S., Clark, A.G., Nadeau, J., McKusick, V.A., Zinder, N., Levine, A.J., Roberts, R.J., Simon, M., Slayman, C., Hunkapiller, M., Bolanos, R., Delcher, A., Dew, I., Fasulo, D., Flanigan, M., Florea, L., Halpern, A., Hannenhalli, S., Kravitz, S., Levy, S., Mobarry, C., Reinert, K., Remington, K., Abu-Threideh, J., Beasley, E., Biddick, K., Bonazzi, V., Brandon, R., Cargill, M., Chandramouliswaran, I., Charlab, R., Chaturvedi, K., Deng, Z., Di Francesco, V., Dunn, P., Eilbeck, K., Evangelista, C., Gabrielian, A.E., Gan, W., Ge, W., Gong, F., Gu, Z., Guan, P., Heiman, T.J., Higgins, M.E., Ji, R.R., Ke, Z., Ketchum, K.A., Lai, Z., Lei, Y., Li, Z., Li, J., Liang, Y., Lin, X., Lu, F., Merkulov, G.V., Milshina, N., Moore, H.M., Naik, A.K., Narayan, V.A., Neelam, B., Nusskern, D., Rusch, D.B., Salzberg, S., Shao, W., Shue, B., Sun, J., Wang, Z., Wang, A., Wang, X., Wang, J., Wei, M., Wides, R., Xiao, C., Yan, C., Yao, A., Ye, J., Zhan, M., Zhang, W., Zhang, H., Zhao, Q., Zheng, L., Zhong, F., Zhong, W., Zhu, S., Zhao, S., Gilbert, D., Baumhueter, S., Spier, G., Carter, C., Cravchik, A., Woodage, T., Ali, F., An, H., Awe, A., Baldwin, D., Baden, H., Barnstead, M., Barrow, I., Beeson, K., Busam, D., Carver, A., Center, A., Cheng, M.L., Curry, L., Danaher, S., Davenport, L., Desilets, R., Dietz, S., Dodson, K., Doup, L., Ferriera, S., Garg, N., Gluecksmann, A., Hart, B., Haynes, J., Haynes, C., Heiner, C., Hladun, S., Hostin, D., Houck, J., Howland, T., Ibegwam, C., Johnson, J., Kalush, F., Kline, L., Koduru, S., Love, A., Mann, F., May, D., McCawley, S., McIntosh, T., McMullen, I., Moy, M., Moy, L., Murphy, B., Nelson, K., Pfannkoch, C., Pratts, E., Puri, V., Qureshi, H., Reardon, M., Rodriguez, R., Rogers, Y.H., Romblad, D., Ruhfel, B., Scott, R., Sitter, C., Smallwood, M., Stewart, E., Strong, R., Suh, E., Thomas, R., Tint, N.N., Tse, S., Vech, C., Wang, G., Wetter, J., Williams, S., Williams, M., Windsor, S., Winn-Deen, E., Wolfe, K., Zaveri, J., Zaveri, K., Abril, J.F., Guigo, R., Campbell, M.J., Sjolander, K.V., Karlak, B., Kejariwal, A., Mi, H., Lazareva, B., Hatton, T., Narechania, A., Diemer, K., Muruganujan, A., Guo, N., Sato, S., Bafna, V., Istrail, S., Lippert, R., Schwartz, R., Walenz, B., Yooseph, S., Allen, D., Basu, A., Baxendale, J., Blick, L., Caminha, M., Carnes-Stine, J., Caulk, P., Chiang, Y.H., Coyne, M., Dahlke, C., Mays, A., Dombroski, M., Donnelly, M., Ely, D., Esparham, S., Fosler, C., Gire, H., Glanowski, S., Glasser, K., Glodek, A., Gorokhov, M., Graham, K., Gropman, B., Harris, M., Heil, J., Henderson, S., Hoover, J., Jennings, D., Jordan, C., Jordan, J., Kasha, J., Kagan, L., Kraft, C., Levitsky, A., Lewis, M., Liu, X., Lopez, J., Ma, D., Majoros, W., McDaniel, J., Murphy, S., Newman, M., Nguyen, T., Nguyen, N., Nodell, M., Pan, S., Peck, J., Peterson, M., Rowe, W., Sanders, R., Scott, J., Simpson, M., Smith, T., Sprague, A., Stockwell, T., Turner, R., Venter, E., Wang, M., Wen, M., Wu, D., Wu, M., Xia, A., Zandieh, A. and Zhu, X. (2001). The Sequence of the Human Genome. Science 291: 1304-1351. Vettore, A.L., da Silva, F.R., Kemper, E.L. and Arruda, P. (2001). The libraries that made SUCEST. Gen. Mol. Biol. 24: 1-7. Warrington, J.A., Nair, A., Mahadevappa, M. and Tsyganskaya, M. (2000). Comparison of human adult and fetal expression and identification of 535 housekeeping/maintenance genes. Physiol. Genomics 2: 143-147. Wiemann, S., Weil, B., Wellenreuther, R., Gassenhuber, J., Glassl, S., Ansorge, W., Bocher, M., Blocker, H., Bauersachs, S., Blum, H., Lauber, J., Dusterhoft, A., Beyer, A., Kohrer, K., Strack, N., Mewes, H.W., Ottenwalder, B., Obermaier, B., Tampe, J., Heubner, D., Wambutt, R., Korn, B., Klein, M. and Poustka, A. (2001). Toward a catalog of human genes and proteins: sequencing and analysis of 500 novel complete protein coding human cDNAs. Genome Res. 11: 422-435. Yao, J., Chiba, T., Sakai, J., Hirose, K., Yamamoto, M., Hada, A., Kuramoto, K., Higuchi, K. and Mori, M. (2004). Mouse testis transcriptome revealed using serial analysis of gene expression. Mamm. Genome 15: 433-451. Yelin, R., Dahary, D., Sorek, R., Levanon, E.Y., Goldstein, O., Shoshan, A., Diber, A., Biton, S., Tamir, Y., Khosravi, R., Nemzer, S., Pinner, E., Walach, S., Bernstein, J., Savitsky, K. and Rotman, G. (2003). Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 21: 379-386. |
|