![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
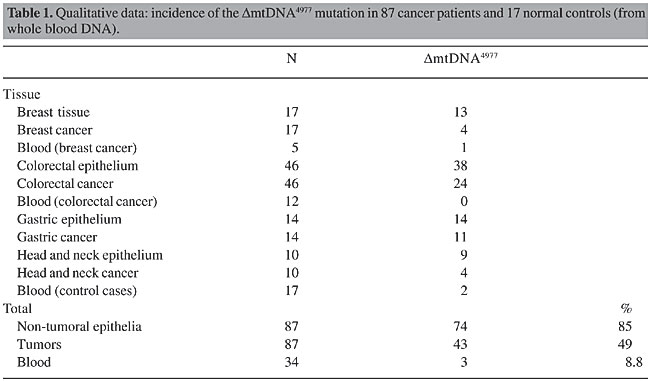
ABSTRACT. Levels of mtDNA4977 deletions (DmtDNA4977) have been found to be lower in tumors than in adjacent non-tumoral tissues. In 87 cancer patients, DmtDNA4977 was detected by multiplex polymerase chain reaction (PCR) amplification in 43 (49%) of the tumors and in 74 (85%) of the samples of non-tumoral tissues that were adjacent to the tumors. DmtDNA4977 deletions were detected in 24% of the breast tumors, 52% of the colorectal tumors, 79% of the gastric tumors, and 40% of the head and neck tumors as compared with 77, 83, 100, and 90% of the adjacent respective non-tumoral tissues at the same DNA template dilution. Based on limiting dilution PCR of 16 tumors and their adjacent non-tumoral tissues, it was found that the amount of DmtDNA4977 was 10- to 100-fold lower in the tumor than in the respective control non-tumoral tissues. Real-time PCR experiments were performed to quantify the number of DmtDNA4977 deletions per cell, by determining the mitochondrial-to-nuclear DNA ratio. In all of the cases of breast, colorectal, gastric, and head and neck cancer the proportion of DmtDNA4977 in tumors was lower than that of the respective non-tumoral tissue. Traces of DmtDNA4977 in tumors were apparently due to contamination of tumor tissue with surrounding non-tumoral tissue, as evidenced by tumor microdissection and in situ PCR techniques, suggesting that tumors are essentially free of this mutation. Although the metabolic effect of DmtDNA4977 may be minimal in normal (non-tumor) tissue, in tissue under stress, such as in tumors, even low levels of DmtDNA4977 deletions may be intolerable. Key words: Mitochondria, Mitochondrial DNA, In situ PCR, Cancer, Kerns-Sayre syndrome deletion mutation, DmtDNA4977, Real-time PCR INTRODUCTION Mitochondria and mitochondrial DNA (mtDNA) have been involved in the modulation of the tumorigenic phenotype (Soslau et al., 1974; Pederson, 1978; Wilkie and Evans, 1982; Israel and Schaeffer, 1987, 1988; Hayashi et al., 1992; Baggetto, 1992; Morais et al., 1994; Bianchi et al., 1995; Liang and Hays, 1996; Cavalli et al., 1997; Dani et al., 2003). Mitochondria are the main energy source of the cell, and the role of mitochondria in the bioenergetics of cancer cells has been reviewed (Pederson, 1978; Bianchi et al., 1995; Liang and Hays, 1996; Dani et al., 2003). Human mitochondrial DNA, a 16,569-bp circular double-stranded molecule, encodes 13 subunits of the respiratory chain complexes, 2 ribosomal RNAs and 22 transfer RNAs. Each nucleated human cell contains a few thousand copies of mtDNA, the somatic mutation rate of which is presumed to be 10 to 20 times higher than that of nuclear DNA (Shenkar et al., 1996). The most common somatic mtDNA mutation, and also the most often assayed, DmtDNA4977, is a deletion that occurs between nucleotides 8,470 and 13,477 of the human mtDNA; it encompasses five tRNA genes and seven genes encoding sub-units of cytochrome c oxidase, complex I and ATPases. This mutation creates an mtDNA molecule that is smaller than the normal mtDNA molecule, though it is still capable of replication. Being smaller and replication competent, the DmtDNA4977 molecule may accumulate with age, primarily in postmitotic tissues (Cortopassi and Arnheim, 1990; Corral-Debrinski et al., 1991; Hattori et al., 1991; Yen et al., 1991; Cortopassi et al., 1992; Zhang et al., 1992), at varying rates, depending on environmental and genetic factors, such as the mutation rate, the initial frequency of deletions present at conception, and selective factors that affect deleted molecules. In a study of the level of DmtDNA4977 in bronchoalveolar tissues from smokers and non-smokers (Ballinger et al., 1996), a seven-fold higher frequency of this deletion was found in smokers. It is generally assumed that damage to mtDNA inhibits oxidative phosphorylation (OXPHOS) and can cause or contribute to chronic disease. It appears, however, that the situation under proliferative conditions, including carcinogenesis, is completely different. For example, the finding of DmtDNA4977 in hematologically normal individuals, and its absence in proliferative hematological disorders, have been explained by selection against self-renewing stem cells that harbor this deletion (Gattermann et al., 1995). In addition, the frequency of PCR-based detection of DmtDNA4977 has been found to be lower in thyroid, renal, and liver tumors, than in respective non-tumoral tissues (Tallini et al., 1994; Fukushima et al., 1995). We examined the hypothesis that DmtDNA4977 is less frequent and less abundant in different tumor types as compared with non-tumoral adjacent tissue, suggesting that its absence is required for neoplastic growth in these tissues. MATERIAL AND METHODS We screened breast, colorectal, gastric and head and neck tumors, and adjacent non-tumoral tissues for the presence of DmtDNA4977, utilizing multiplex PCR amplification of the new sequence created on the mtDNA molecule by the DmtDNA4977 deletion. To compare the frequency of this deletion in tumors and the adjacent respective non-tumoral epithelial tissues, we performed limiting dilution multiplex PCR, using primer pairs for the common deletion, total mtDNA, and the b-globin gene for normalization, i.e., determination of the mitochondrial-to-nuclear DNA ratio. We also performed multiplex real-time PCR experiments to determine the number of deletions/cell in tumors and in adjacent non-tumoral tissues. Sample preparation A total of 208 fresh-tissue samples, including blood from 17 normal individuals (used as controls) and from 17 cancer patients (12 colorectal and 5 breast), and 87 tissue sample pairs, from both non-tumoral and tumor tissues from cancer patients (17 breast, 46 colorectal epithelium, 14 gastric epithelium, 10 head and neck epithelium) were analyzed for total mtDNA and for the DmtDNA4977 mutation. The 87 non-tumoral tissues and their respective primary tumors and blood samples were from surgical operations performed at the A.C. Camargo Hospital. Twenty-four of 87 tumor samples were microdissected to decrease the contamination of the tumor with surrounding non-tumoral tissue. Total DNA was extracted using the phenol-chloroform method, as described elsewhere (Maniatis, 1989), or with the Nucleon HT® kit for DNA extraction from hard tissue (Amersham Life Science, USA). Absorbances were read at 260 nm and 280 nm to determine DNA purity and concentration. The DNA was diluted to 50 ng/µl and stored at -20°C. Multiplex PCR for the amplification of total mtDNA, the DmtDNA4977 mutation and a nuclear gene for normalization (determination of mitochondrial-to-nuclear DNA ratio) Multiplex PCRs were carried out with three primer pairs: ND6A/ND6B and HSAS8542/HSSN8416 (Zullo, 2002) and GH20/PCO4 (Saiki et al., 1988). Primer ND6A, located at bp 13,988-14,007 (5’-TTC TCC TAG ACC TAA CCT GA-3’) of the mtDNA and primer ND6B, located at bp 14,472-14,453 (5’-GGA TAT ACT ACA GCG ATG GC- 3’) of the mtDNA, were used to amplify a 485-bp fragment in a rarely deleted region containing subunit 6 of the NADH gene, as a measure of total mtDNA in each sample. Primers HSAS8542 (5’-TGT GGT CTT TGG AGT AGA AAC C-3’) and HSSN8416 (5’-CCT TAC ACT ATT CCT CAT CAC C-3’) were used to amplify a 127-bp region created by the DmtDNA4977 deletion. Primers GH20 (5’-GAA GAG CCA AGG ACA GGT AC-3’) and PCO4 (5’-CAA CTT CAT CCA CGT TCA CC-3’) were used to amplify a 268-bp region from the b-globin gene, for normalization of total mtDNA copy numbers as a function of the total copy numbers of a nuclear gene. PCR reactions were carried out in a 25-µl reaction volume with 0.125 µg genomic DNA, 2.5 x 10-2 U/µl Ampli Taq GoldTM, 2.5 mM MgCl2, 0.175 mM each dNTP, and 1.0 µM of each primer. The enzyme Ampli Taq GoldTM (Perkin Elmer) was activated through a pre-PCR heat step of 10 min at 95°C. After that, the reaction mixture was cycled 35 times at 95°C for 30 s, 60°C for 30 s and 72°C for 45 s. PCRs were performed in duplicate in an MJ Research Inc. PTC-100TM thermocycler. Ampli Taq GoldTM, buffer and MgCl2 were from Perkin Elmer, dNTPs were from Promega and the oligonucleotides were synthesized at the Ludwig Institute for Cancer Research using a 391-DNA Synthesizer-PCR Mate-Applied Biosystems. Limiting dilution experiments To test the reproducibility and sensibility of the methodology, amplicons corresponding to the 127-bp region created by the DmtDNA deletion, the 485 bp of the rarely deleted region of the mtDNA, and the 268 bp of the b-globin gene, were cloned and sequenced. After quantification, plasmids were diluted to a final concentration of 5 pg/µl each and were used as templates, separately or combined in equal concentrations in limiting dilution multiplex PCRs. To compare the proportion of deleted mtDNA/total mtDNA in 16 tumors and in their non-tumoral adjacent tissues, which were selected at random, PCRs were carried out using serial dilutions of DNA template (10-1 to 10-8), with the three primer pairs and the same PCR conditions as described above. Electrophoresis PCR products were analyzed on 8% polyacrylamide gels in TBE buffer, pH 8.3. Six microliters of the reaction products was mixed with tracing dye (bromophenol blue, xylene cyanol and Ficoll), loaded on gels and run for 30 min at 150V. DNA was stained with silver nitrate (Sanguinetti et al., 1994) and the gels were photographed (Figures 1 and 2). Cloning and sequencing PCR products containing the total mtDNA fragments (127 bp and 485 bp), the b-globin gene (268 bp), the b-actin gene (295 bp), and the sequence created by the common deletion (127 bp) were cloned into pUC18 vectors using the Sure Clone® Ligation Kit (Pharmacia-Biotech, Sweden) and were purified from bacteria using the Wizard® Miniprep Purification System (Promega, USA). Sequencing was performed in an ABI Prism®377 (Perkin-Elmer, USA). These plasmids were used as controls in multiplex PCRs, multiplex limiting dilution PCRs and real-time PCRs. Quantitative real-time PCR For real-time PCR quantifications, nine tumor/non-tumoral tissue pairs were selected at random for real-time PCR quantifications. We ran two multiplex real-time PCRs for each tumor and its non-tumoral adjacent tissue DNA, both in triplicate: (i) the first multiplex real-time PCR amplified total mtDNA and DmtDNA4977, simultaneously, for quantification of the number of deleted mtDNA molecules per total mtDNA molecules (DmtDNA4977/total mtDNA); (ii) the second multiplex real-time PCR was performed for the simultaneous quantification of total mtDNA and genomic DNA (b-actin), giving an estimate of mtDNA molecules per cell (total mtDNA/cell). By multiplying the two factors, (i) x (ii), we obtained an estimate of the number of DmtDNA4977 deletions per cell in each tumor and non-tumoral sample. Real-time amplifications were carried out using two different PCR mixes, each one containing two primer pairs and two labeled probes: mix-1 contained primer pair HSS1307 (5'-gta ccc acg taa aga cgt tag g-3')/HSAS1433 (5'-tac tgc taa atc cac ctt cg-3') [24] (for the amplification of a rarely deleted 127-bp region of the mtDNA that includes the 12S-rRNA gene, as an estimate of total mtDNA copies), primer pair HSAS8542/HSSN8416 (region created by the DmtDNA deletion, as described above), DmtDNA probe (5’FAM-TGG CAG CCT AGC ATT AGC AGG-TAMRA3’) and total mtDNA probe (5’TET-CCC ATG AGG TGG CAA GAA AT-TAMRA3’); mix-2 contained primer pair b-actin forward/b-actin reverse (included in the TaqMan® PCR Core Reagent kit, Perkin, USA, amplifying a 295-bp fragment of the b-actin gene), primer pair HSS1307/HSAS1433 (as described above), b-actin probe (included in the TaqMan® PCR core reagent kit, Perkin, USA) and total mtDNA probe (as described above). The probes labeled with both a fluorescent reporter dye, and a quencher dye, were synthesized by PE-Applied Biosystems. TaqMan® reactions were prepared in a 96-well plate using 100 ng genomic DNA, 200 nM of each labeled probe, 300 nM of each primer, 3.5 mM MgCl2, 100 nM of each dNTP, except dUTP (200 nM), 0.01 U/µl AmpErase UNG, and 0.025 U/µl Ampli Taq GoldTM. Samples were cycled through 94°C for 15 s and 60°C for 1 min, for a total of 40 cycles and emitted fluorescence, which was detected over the course of the run in an ABI PRISM 7700 Sequence Detection System (PE-Applied Biosystems, USA). In situ PCR For in situ PCR experiments, pairs of formalin-fixed, paraffin-embedded tumor/non-tumoral tissue slices were mounted on clean, sylanized glass slides. Following deparaffinization and rehydration, the tissue slices were incubated for 15 min in a DMSO-diluted MitoTracker Green FM (a mitochondria selective probe from Molecular Probes, Inc., USA) and washed in SSC buffer. In situ PCR was carried out using the DmtDNA4977 mix, containing primer pair HSAS8542/HSSN8416 (as described above) and the DmtDNA4977 probe labeled with TRITC (Martínez et al., 2001). Negative controls (no primers in the reaction mixture) were run in parallel. The in situ PCR mixture was applied directly onto the slices at 50 µl/slice and was covered with glass slips fixed in place with rubber cement. The slides were placed in the reaction block of a Hybaid Omnislide PCR machine (Hybaid, UK). At the end of the in situ PCR, the rubber cement and cover slips were carefully removed and the slides were quickly washed in a 2X SSC buffer, followed by a 5-min incubation in a DAPI solution to counterstain the nuclei. The glass slides were washed in a shaking water bath for 5 min, dehydrated in increasing alcohol concentrations, air dried and mounted with cover slips and anti-fading media. They were examined under a fluorescence microscope (Zeiss, Germany). RESULTS The frequency of DmtDNA4977 deletions was lower in tumors than in adjacent non-tumoral tissues (P < 0,001, Table 1). The mutations were detected in fewer of the breast, colorectal, gastric and head and neck tumors than in the adjacent respective non-tumoral tissues, at the same template DNA dilutions (Table 1).
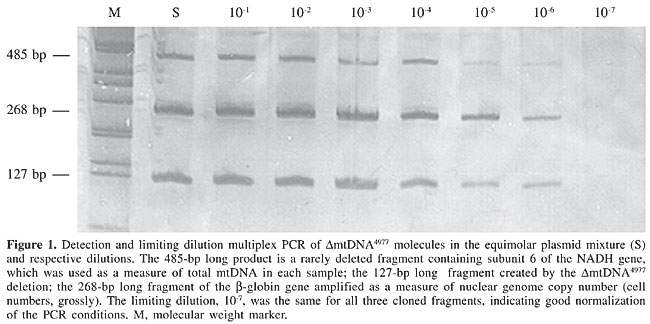
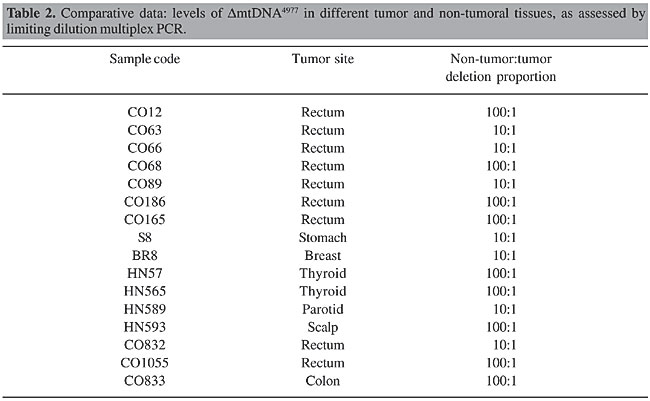
Limiting dilution multiplex PCRs were carried out using mixed equimolar concentrations of plasmids containing fragments of the region created by the DmtDNA deletion, total mtDNA and the b-globin gene (Figure 1). Despite the differences observed in the intensities of the bands, which were probably due to competition favoring smaller fragments, all three fragments were amplified up until the same limiting dilution, 10-6. When we compared the frequency of the deletions in 16 tumors, and in the adjacent respective non-tumoral epithelial tissues, by limiting dilution multiplex PCR, we found 10- to 100-fold less copies of DmtDNA4977 in tumors than in the respective adjacent non-tumoral tissues (P < 0.001, Table 2). In none of the cases of breast, colorectal, gastric or head and neck cancer did the frequency of the DmtDNA4977 deletion in tumors exceed that of the respective non-tumoral tissue.
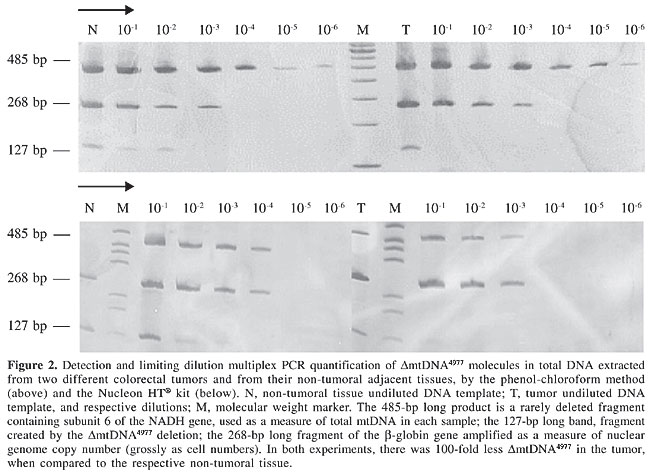
For example, DNA was extracted from two different colorectal tumors and from non-tumoral adjacent tissues, by the phenol-chloroform method and the Nucleon HT® kit, respectively; it was then amplified by limiting dilution multiplex PCR (Figure 2). In both cases, DmtDNA4977 proportions were 100-fold higher in the non-tumoral tissues.
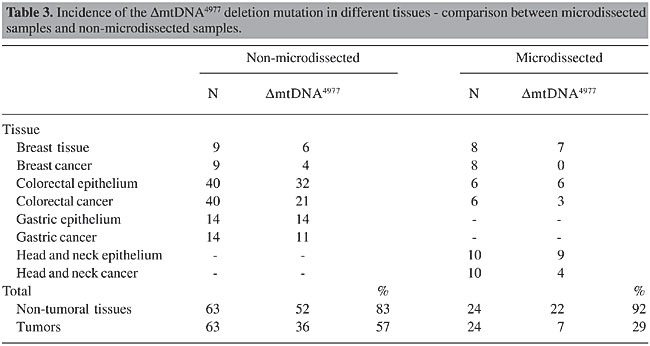
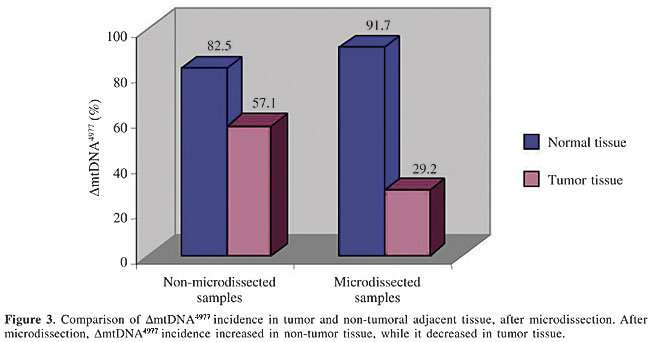
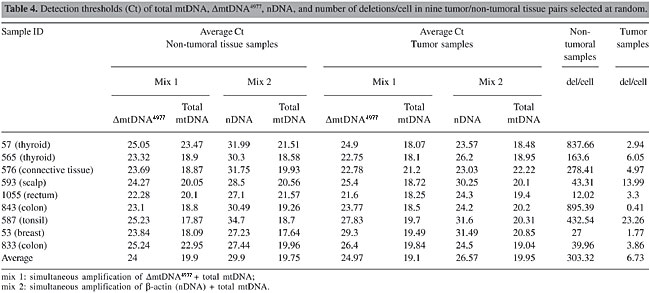
DmtDNA4977 was detected in 57% of the non-microdissected tumor samples, whereas in microdissected samples this percentage decreased to 29% (Table 3 and Figure 3, P < 0.001). The results of real-time PCR experiments are illustrated in Figure 4. The number of deletions/cell in 9 tumors and in the respective non-tumoral tissues were significantly different (P < 0.001, Table 4).
In the in situ PCR experiments, mutation spots were seen in non-tumoral epithelial cells, but not in tumor cells (Figure 5).
DISCUSSION In addition to postmitotic tissues, non-tumoral tissues have also been found to accumulate DmtDNA4977 deletions, though neoplastic transformation in a variety of tissues, such as thyroid and kidney (Tallini et al., 1994), liver (Fukushima et al., 1995), and breast, colorectal and gastric epithelia, appears to involve enrichment of these tissues with cells that do not carry the DmtDNA4977 mutation. This phenomenon has also been described in blood and bone marrow of hematologically normal adults undergoing sternotomy for cardiac surgery and in patients with myelodysplastic syndromes (Gattermann et al., 1995). DmtDNA4977 deletions were detected in 35% of the normal adults, though they were not detected in patients with myelodysplastic syndromes, acute myeloid leukemia or chronic myeloid leukemia, suggesting selection under circumstances of proliferative stress (Gattermann et al., 1995). We observed the lowest incidence of DmtDNA4977 in blood (8.8%); these positive blood samples corresponded to two kidney-transplanted patients and one breast cancer patient. Two of our positive blood samples were from patients undergoing surgical operations, so it is possible that the surgical operation is a factor that affects the detectability of the DmtDNA4977, perhaps by freeing DmtDNA4977 molecules from tissues into the blood stream (Fliss et al., 2000). We observed a trend towards a higher accumulation of the DmtDNA4977 molecule in non-tumoral tissues, when compared to the respective cancers (Table 1). The frequency of the DmtDNA4977 mutation was high in non-tumoral breast, colorectal and head and neck tissues and relatively low in breast, colorectal and head and neck tumors (Table 1). The frequency of the mutation was even higher in gastric tumors, than in colorectal, breast and head and neck tumors, though the trend towards a lower frequency of this deletion in tumors compared to adjacent respective non-tumoral tissues still persisted in the gastric epithelium (Table 1). Moreover, in all of the breast, colorectal, gastric and head and neck tumor samples the proportion of the DmtDNA4977 deletion in tumors was lower than that of the respective non-tumoral tissues (Table 2). Real-time PCR run in the ABI Prism 7700 sequence detection system is a very sensitive technique to measure fine distinctions in the frequency of deletions. A recent report (Chabi et al., 2003) indicates that quantitative real-time PCR has a higher sensitivity than Southern blot analysis in the quantification of mtDNA. Our real-time PCR results are consistent with our limiting dilution PCR results, confirming that non-tumoral tissues contain more deletions than tumor tissues, and that the difference is numerically significant. One possible explanation for the differences found between different non-tumoral tissues is the existence of tissue-specific mtDNA turnover rates and various environmental and genetic influences, e.g., variation between tissues in the expression of nuclear genes that encode mitochondrial functions (Wallace, 1993). One explanation for the presence of DmtDNA4977 in some tumor samples, though clearly at lower concentrations than in the respective non-tumoral tissues, may be the inevitable contamination of the tumor sample with surrounding non-tumoral tissue (e.g., epithelial, vascular, and connective tissues). Support for this explanation is granted by our finding that contamination, i.e., detection of DmtDNA4977 from non-tumoral tissue in tumors, was reduced by microdissection of the tumor tissue (Table 3). In situ PCR provided additional support (Figure 5). One major point that needs to be addressed is the question of whether the low levels of DmtDNA4977 found in non-tumoral tissue have any detectable metabolic consequence. Possibly, although the metabolic effect may be minimal in normal (non-tumor) tissue, in tissue under stress, such as in tumor-transformed cells, even low frequencies of mtDNA4977 deletions are intolerable. Heteroplasmy for the DmtDNA4977 mutation in the tumor could be tolerated at some stage of the development of a tumor, assuming that heteroplasmy for DmtDNA4977 is acceptable until a given threshold is reached. However, the extremely low frequency of the DmtDNA4977 mutation in non-tumoral tissues (less than 0.01% of total mtDNA copy numbers) suggests that this threshold is very low indeed. We speculate that once this threshold is achieved, the affected cell dies rapidly. It is conceivable that this threshold is higher for non-tumoral cells than for tumor cells. This would explain why DmtDNA4977 mutations accumulate to detectable levels in non-tumoral tissues, but not in tumors. It is unlikely that an intracellular selection operates to progressively purge the tumor from DmtDNA4977-positive cells. Rather, DmtDNA4977-positive cells would never develop into a tumor. This is in contrast with the progressive accumulation, in tumors, of less critical mtDNA mutations, such as those affecting hypervariable regions in the mtDNA molecule, which have been reported in colorectal and gastric tumors (Burgart et al., 1995; Alonso et al., 1997). The DmtDNA4977 deletion, which affects important genes involved in OXPHOS, such as ATPase 6, ATPase 8, cytochrome oxidase III, and NADH subunits ND3, ND4, ND4L, and ND5, may have a strong metabolic disadvantage, so that cells carrying this mutation are selected against, and as a consequence the DmtDNA4977 mutation is apparently selected against in tumors derived from epithelia that are initially heteroplasmic for this mutation. The selection force is reflected in a 10- to 100-fold decrease in the DmtDNA4977: total mtDNA ratio in the tumor mass, when compared to the adjacent respective non-tumoral tissue. It is as yet not clear whether DmtDNA4977 offers any protection against cancer; however, given the decreased incidence of such detections in tumor tissues, DmtDNA4977 appears to be important enough to be selected against. Although less than 0.01% mtDNA4977 deletions among the total mtDNA, may be tolerated in normal (non-tumor) tissue, in tissue under stress, such as in tumor-transformed cells, even such low levels of mtDNA4977 deletions may be intolerable. One could argue that tumor cells do not rely entirely on OXPHOS to survive, and hence that mutations such as DmtDNA4977 would not be metabolically detrimental. It is well known from the pioneer work of Warburg (1956) that tumor cells grown in culture have a higher capacity for glycolysis than does non-tumor tissue. However, this higher glycolytic capacity may be due to increased growth and nutrient demands of tumor cells, which is not an argument against the importance of OXPHOS for the survival and growth of tumors. It is precisely because of higher nutrient demands, including oxygen demand, that many tumors continue to grow in vivo because they induce neovascularization (Ausprunk et al., 1975; Brooks et al., 1994). In the embryonic egg model system, for example, it has been found that tumor growth has avascular and vascular phases, depending on tumor age and size (Knighton et al., 1977). Under the more exacting in vivo circumstances, one must concede that OXPHOS coexists with glycolysis, and the best adapted cells, which are also the most rapidly proliferating cells, would be precisely those capable of both OXPHOS and glycolysis. In support of this conclusion is the evidence that depletion of mitochondrial DNA diminishes the tumorigenic phenotype (Israel and Schaeffer, 1987, 1988; Hayashi et al., 1992; Cavalli et al., 1997), indicating that mitochondrially encoded functions are essential for the maintenance of viable tumor cells. The somatic accumulation of the DmtDNA4977 mutation in non-tumoral tissues with age, and the apparent absence of this mutation in neoplastic tissues suggest that the mitochondrial dysfunction caused by the DmtDNA4977 deletion greatly affects the survival of proliferating tumor cells. CONCLUSION We conclude that the DmtDNA4977 mutation confers a metabolic disadvantage to proliferating cells and thus is selected against in the highly proliferative tumor tissue. The selection pressure is presumably relaxed in the non-tumoral tissue with a lower proliferative rate, thus allowing the accumulation of the DmtDNA4977 mutation at varying levels in aging non-tumoral tissue. ACKNOWLEDGMENTS We thank Cristiane Ayres and Adriana A. Marques for DNA sequencing. Dr. Otavia L. Caballero, Valéria Paixão and Carlos helped with the preparation of samples. We also thank Drs. Carl Merril and Steven Zullo for their comments on the manuscript. M.A.C. Dani and S.U. Dani were recipients of FAPESP fellowships - the São Paulo State Research Support Foundation. Research supported by FAPESP, the Ludwig Institute for Cancer Research and Genon Ltda. REFERENCES Alonso, A., Martin, P., Albarran, C., Aguilera, B., Garcia, O., Guzman, A., Oliva, H. and Sancho, M. (1997). Detection of somatic mutations in the mitochondrial DNA control region of colorectal and gastric tumors by heteroduplex and single-strand conformation analysis. Electrophoresis 18: 682-685. Ausprunk, D.H., Knigthon, D.R. and Folkman, J. (1975). Vascularization of normal and neoplastic tissues grafted to the chick chorioallantois. Role of host and preexisting graft blood vessels. Am. J. Pathol. 79: 597-628. Baggetto, L.G. (1992). Role of mitochondria in carcinogenesis. Eur. J. Cancer 29A: 156-159. Ballinger, S.W., Bouder, T.G., Davis, G.D., Judice, S.A., Nicklas, J.A. and Albertini, R.J. (1996). Mitochondrial genome damage associated with cigarette smoking. Cancer Res. 56: 5692-5697. Bianchi, M.S., Bianchi, N.O. and Bailiet, G. (1995). Mitochondrial DNA mutations in normal and tumor tissue from breast cancer patients. Cytogenet. Cell Genet. 71: 99-103. Brooks, P.C., Montgomery, A.M.P., Rosenfeld, M., Reisfeld, A., Tianhua, H., Klier, G. and Cheresh, D.A. (1994). Integrin avb3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 79: 1115-1164. Burgart, L.J., Zheng, J., Shu, Q., Strickler, J.G. and Shibata, D. (1995). Somatic mitochondrial mutation in gastric cancer. Am. J. Pathol. 147: 1105-1111. Cavalli, L.R., Varella-Garcia, M. and Liang, B.C. (1997). Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 8: 1189-1198. Chabi, B., Mousson De Camaret, B., Duborjal, H., Issartel, J.P. and Stepien, G. (2003). Quantification of mitochondrial DNA deletion, depletion, and overreplication: application to diagnosis. Clin. Chem. 49: 1309-1317. Corral-Debrinski, M., Stepian, G., Shoffner, J.M., Lott, M.T., Kanter, K. and Wallace, D.C. (1991). Hypoxemia is associated with mitochondrial DNA damage and gene induction: implications for cardiac disease. J.A.M.A. 266: 1812-1816. Cortopassi, G.A. and Arnheim, N. (1990). Detection of a specific mitochondrial DNA deletion in tissues of older individuals. Nucleic Acids Res. 18: 6927-6933. Cortopassi, G.A., Shibata, D., Soong, N.-W. and Arnheim, N. (1992). A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 89: 7370-7374. Dani, S.U., Dani, M.A.C. and Simpson, A.J.G. (2003). The common mitochondrial DNA deletion DmtDNA4977: shedding new light on the concept of a tumor suppressor mutation. Med. Hypotheses 61: 60-63. Fliss, M.S., Usadel, H., Caballero, O.L., Wu, L., Buta, M.R., Eleff, S.M., Jen, J. and Sidransky, D. (2000). Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 287: 2017-2019. Fukushima, S., Honda, K., Awane, M., Yamamoto, E., Takeda, R., Kaneko, I., Tanaka, A., Morimoto, T., Tanaka, K. and Yamaoka, Y. (1995). The frequency of 4977 base pair deletion of mitochondrial DNA in various types of liver disease and in normal liver. Hepatology 21: 1547-1551. Gattermann, N., Berneburg, M., Heinisch, J., Aul, C. and Schneider, W. (1995). Detection of ageing-associated 5-kb common deletion of mitochondrial DNA in blood and bone marrow of hematologically normal adults. Absence of the deletion in clonal bone marrow disorders. Leukemia 9: 1704-1710. Hattori, K., Tanaka, M., Sugiyama, S., Obayashi, T., Ito, T., Satake, T., Hanaki, Y., Asai, J., Nagano, M. and Ozawa, T. (1991). Age dependent increase in deleted mitochondrial DNA in the human heart: possible contributory factor to presbycardia. Am. Heart J. 121: 1735-1742. Hayashi, J.-I., Takemitsu, M. and Nonaka, I. (1992). Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa cells by introduction of mitochondrial DNA from normal human cells. Somat. Cell Mol. Genet. 18: 123-129. Israel, B.A. and Schaeffer, W.I. (1987). Cytoplasmic suppression of malignancy. In Vitro Cell. Dev. Biol. 23: 627-632. Israel, B.A. and Schaeffer, W.I. (1988). Cytoplasmic mediation of malignancy. In Vitro Cell. Dev. Biol. 24: 487-497. Knighton, D., Ausprunk, D., Tapper, D. and Folkman, J. (1977). Avascular and vascular phase of tumor growth in the chick embryo. Br. J. Cancer 35: 347-356. Liang, B.C. and Hays, L. (1996). Mitochondrial DNA copy number changes in human gliomas. Cancer Lett. 105: 167-173. Maniatis, T., Sambrook, J. and Fritsch, E.F. (1989). Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA. Martínez, A., Man, Y.G., Zullo, S.J. and Cuttitta, F. (2001). In situ amplification and detection of nucleic acids. In: Immunocytochemistry and In situ Hybridization in the Biomedical Sciences (Beesley, J., ed.). Birkhäuser, Boston, MA, USA, pp. 156-174. Morais, R., Zinkewich-Pootti, K., Parent, M., Wang, H., Babai, F. and Zollinger, M. (1994). Tumor forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA. Cancer Res. 54: 3889-3896. Pederson, P.L. (1978). Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor Res. 22: 198-274. Saiki, R.K., Gelfand, D.H., Stoffel, S., Scharf, S.J., Higuchi, R., Horn, G.T., Mullis, K.B. and Erlich, H.A. (1988). Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239: 487-491. Sanguinetti, C.J., Dias Neto, E. and Simpson, A.J.G. (1994). Rapid silver staining and recovery of PCR products separated on polyacrylamide gels. Biotechniques 17: 915-919. Shenkar, R., Navidi, W., Tavaré, S., Dang, H.M., Chomyn, G.A., Cortopassi, G. and Arnheim, N. (1996). The mutation rate of the human mtDNA deletion mtDNA4977. Am. J. Hum. Genet. 50: 772-780. Soslau, G., Fuhrer, J.P., Nass, M.M.K. and Warren, L. (1974). The effects of ethidium bromide on the membrane glycopeptides in control and virus transformed cells. J. Biol. Chem. 249: 3014-3020. Tallini, G., Ladanyi, M., Rosai, J. and Jhanwar, S.C. (1994). Analysis of nuclear and mitochondrial DNA alterations in thyroid and renal oncocytic tumors. Cytogenet. Cell Genet. 66: 253-259. Wallace, D.G. (1993). Mitochondrial diseases: genotype versus phenotype. TIG 9: 128-133. Warburg, O. (1956). On the origin of cancer cells. Science 123: (3191) 309-314. Wilkie, D. and Evans, I. (1982). Mitochondria and the yeast cell surface: Implication for carcinogenesis. Trends Biochem. Sci. 7: 147-151. Yen, T.-C., Sue, J.-H., King, K.-L. and Wei, Y.-H. (1991). Aging associated 5-kb deletion in human liver mitochondrial DNA. Biochem. Biophys. Res. Commun. 178: 124-131. Zhang, C., Baumer, A., Maxwell, R.J., Linnane, A.W. and Nagley, P. (1992). Multiple mtDNA deletions in an elderly individual. FEBS Lett. 297: 34-38. Zullo, S.J. (2002). In situ PCR of the common human mitochondrial DNA deletion: Is it related to apoptosis? Chapter 6. In: Mitochondrial DNA: Methods and Protocols (Copeland, W.C., ed.). Humana Press, Clifton, NJ, USA. |
|