![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
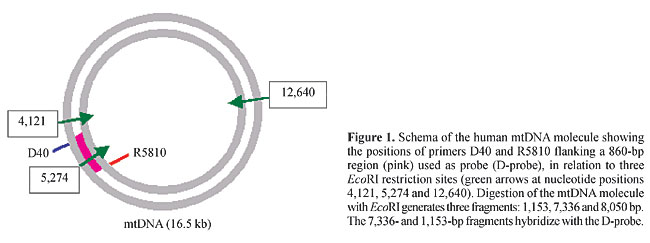
ABSTRACT. We developed, and quantitatively and qualitatively evaluated an easily reproducible method for high yield purification of mitochondrial DNA (mtDNA) from human placentae by mechanical tissue disruption, differential centrifugation of mitochondria, enzymatic digestion, phenol extraction and ethanol precipitation. Average mtDNA yields were 2.5 mg/g tissue (without an RNAse treatment step) and 1.5 mg/g tissue (with an RNAse treatment step). This mtDNA migrated as a 16.5-kb isolated band in agarose gels; it yielded fragments of expected sizes after digestion with restriction enzymes; it successfully served as a template in long PCR for amplification of mtDNA sequences, and hybridized to an mtDNA probe in a predictable fashion. MtDNA yields of this method were 10-fold higher than those of previously reported ones for mtDNA purification from freshly obtained human cells and tissues, with the advantage that more placental tissue can be obtained for mtDNA purification than other types of tissue, at lower cost, and with minimal or no ethical issues. Key words: Mitochondrion, mtDNA purification, Human placenta INTRODUCTION Human mitochondrial DNA (mtDNA) is a self-replicating, 16,569-bp circular double-stranded molecule, localized in mitochondria. Human mtDNA encodes 13 subunits of the respiratory chain complexes, 2 ribosomal RNAs and 22 transfer RNAs. Since the first report of the complete sequence of mtDNA (Anderson et al., 1981), there has been no successful attempt at stably cloning and replicating the full-length mtDNA sequence; the in vitro production of human mtDNA molecules has been hampered by difficulties associated with cloning such a high molecular weight plasmid (Bigger et al., 2000). Isolation of mtDNA from human tissues and cells is the only known source of large amounts of intact mtDNA suitable for a variety of molecular manipulations, including preparation of mtDNA probes, generation and analysis of restriction enzyme fragments, construction of artificial mitochondrial vectors for gene therapy or gene marking purposes, and efficient transfection of mtDNA to isolated mitochondria or cells in culture. Standard methods for mitochondria isolation and mtDNA purification from cells and tissues are relatively high cost, time-consuming, labor-intensive and usually result in low yields of mtDNA, typically 1-3 mg/ml packed cells or 0.05-0.1 mg/g tissue (Drouin, 1980; Hare et al., 1980; King and Attardi, 1988; Ausenda and Chomyn, 1996; Chomyn, 1996). Here we report and evaluate a technique for high yield purification of intact mitochondrial DNA based on a modification of a method of preparation of human placenta mitochondria (Gasnier et al., 1993), followed by standard enzymatic digestion of proteins and phenol-chloroform DNA extraction. While the cell fractionation step avoids contamination of the mtDNA preparation by bulk DNA, the digestion and extraction steps assure the purity of the mtDNA. Although this technique takes 1-2 days, it is a high yield, low cost method. MATERIAL AND METHODS Purification In three independent experiments, a placenta was obtained from the local University Hospital (Hospital das Clínicas de Ribeirão Preto), chilled on ice immediately after cesarean section and transported to the laboratory. All operations were carried out in a cold room (4°C). Each placenta was washed with wash buffer (1 mM EDTA, 50 mM Tris-HCl, pH 7.4), separated from connective tissue with sterile surgical scissors and forceps, cut into small pieces, weighed (180 g), extensively washed in wash buffer, and placed in 250 ml of wash buffer in a 1-liter capacity kitchen blender (2-speed home blender, Walita, Brazil). The placenta was blended at high speed until complete homogenization. This suspension was blended again at high speed five times for 7 s, cooled down for 30 s between each blending time and then transferred to 45-ml polycarbonate tubes, and finally centrifuged in a Sorvall centrifuge at 960 g for 15 min to separate unbroken cells, nuclei, and connective tissue fibers. The supernatant was poured through cheesecloth into a funnel mounted on a beaker, then immediately transferred to new polycarbonate tubes and centrifuged at 8,600 g for 15 min to sediment the mitochondrial fraction. The supernatant was discarded and each mitochondrial pellet was resuspended, with the help of a standard polyethylene disposable bulb pipet, in 30 ml of resuspension buffer (250 mM sucrose, 1 mM EDTA, 10 mM Tris-HCl, pH 7.4) and then sedimented at 8,600 g for 15 min. This step was repeated three more times, for a total of four washes. After the final wash, each mitochondrial pellet was resuspended in 600 ml TE-Tween buffer (50 mM Tris, 1 mM EDTA, 0.5% Tween-20) and transferred to 1.5-ml microcentrifuge tubes containing 100 ml proteinase K (Life Technologies, Inc., Rockville, MD, USA, 20 mg/ml stock) for disruption of the mitochondria. Samples were incubated at 55°C for 3 h and then at 37°C overnight. In a series of experiments, RNAse-A (10 mg/ml stock; Life Technologies, Inc.) was added 1:10 (v/v) to each tube (about 80 ml RNAse/tube), followed by incubation at 37°C for 1 h, before the proteinase K step. After overnight incubation in proteinase K, another 100 ml proteinase K was added to each tube, followed by 2-h incubation at 55°C. To purify mtDNA from proteins and lipids, phenol-chloroform-isoamyl alcohol (25:24:1) was added 1:1 (v/v) to each tube, and mixed by several hand inversions. The samples were centrifuged at 12,000 rpm in a tabletop microcentrifuge, for 1 min. The aqueous phase was transferred to a new 1.5-ml microcentrifuge tube, 10% (v/v) 3 M sodium acetate was added, followed by the addition of 2.5 volumes of ice-cold absolute ethanol and incubation for 20 min at -70°C. Precipitated mtDNA was sedimented in a tabletop microcentrifuge at 12,000 rpm, at 4°C, for 30 min. The pellet was washed once with 70% ethanol, air-dried and resuspended in 100 ml sterile TE buffer (10 mM Tris-HCl, 10 mM EDTA, pH 7.5-8.0). The concentration of the purified mtDNA was determined in a DU-650 spectrophotometer (Beckman, Palo Alto, CA, USA). For long PCR experiments, aliquots of this mtDNA were further chromatographically purified in a glass bead column (GlassMaxä, Life Technologies, Inc.), according to the manufacturer instructions. Restriction endonuclease assay Purified mtDNA was digested with BamHI and HindIII (Life Technologies, Inc.), according to the manufacturer instructions. The products of the digestion were separated on 0.8% agarose gels, and stained with ethidium bromide. PCR assay Multiplexed long PCR amplification of mtDNA target sequences was carried out with the following primers (Reynier and Malthiery, 1995; Dani et al., 1998; Zullo, 2002): ND6-A (5'-TCCTCCTAGACCTAACCTGA-3'), ND6-B (5'-GGATATACTACAGCGATGGC-3'), REagI (5'-CCGCGGCCGTTAAACATGTGTCACTG-3'), dHSSN1307 (5'-GTACCCACGTAAAGACGTTAGG-3'), dHSSN8416 (5'-CCTTACACTATTCCTCATCACC-3'), and R40 (5'-TAAGATTGAGAGAGTGAGGAG-3'). PCR reaction mix was prepared on ice with 300 nM of each primer, 500 mM of each dNTP, PCR buffer with 2.25 mM MgCl2, 100-200 ng template mtDNA, 3.5 U Expandä (Boehringer, Mannheim, Germany) polymerase enzyme mix and double distilled H2O to a final 25 ml reaction volume, and overlaid with a drop of mineral oil. This reaction mix was immediately brought to a Hybaid Expressä thermocycler (Hybaid, Action Court, Middlesex, UK) previously paused at 92°C (hot start technique). Cycling conditions were as follows: 92°C for 2 min; then 10 cycles of 92°C for 10 s, 55°C for 30 s, 68°C for 15 min; then 20 cycles of 92°C for 10 s, 55°C for 30 s with 20 s increment/cycle, 68°C for 15 min; then a final extension at 68°C for 7 min and a hold step at 4°C. Southern blot assay Seven micrograms each of total DNA obtained from surgically extirpated tumor samples and normal tissue adjacent to the tumors, as well as total leukocyte DNA from human peripheral blood and mtDNA purified by the above described method were digested with the restriction enzyme EcoRI, electrophoresed in 1% agarose gels and transferred to nylon membranes (Sigma, St. Louis, MO, USA). The nylon membranes were hybridized with the D-probe, as described below. D-Probe preparation Primers D40 (5'-TCCATCATAGCAGGCAGTTG-3') and R5810 (5'-TACCAGCTCCGAGGTGATTT-3') were used to amplify an 860-bp fragment of the human mtDNA spanning from nucleotide position 4,950 to nucleotide position 5,810. Figure 1 shows the localization of the primers and selected restriction sites on the mtDNA molecule. PCR reactions were carried out in a 25-ml reaction volume containing 100 ng human genomic DNA; 2.5 x 10-2 U/ml Ampli Taq Goldä DNA polymerase (Applied Biosystems, Foster City, CA, USA); 2.5 mM MgCl2; 0.175 mM of each dNTP and 1.0 mM of each primer. Ampli Taq Goldä DNA polymerase was activated through a pre-PCR heat step of 10 min at 95°C. After that, the reaction mixture was cycled 35 times at 95°C for 30 s, 60°C for 30 s and 72°C for 45 s, in a Hybaid Express (PCR-Express, Hybaid, Middlesex, UK) thermocycler.
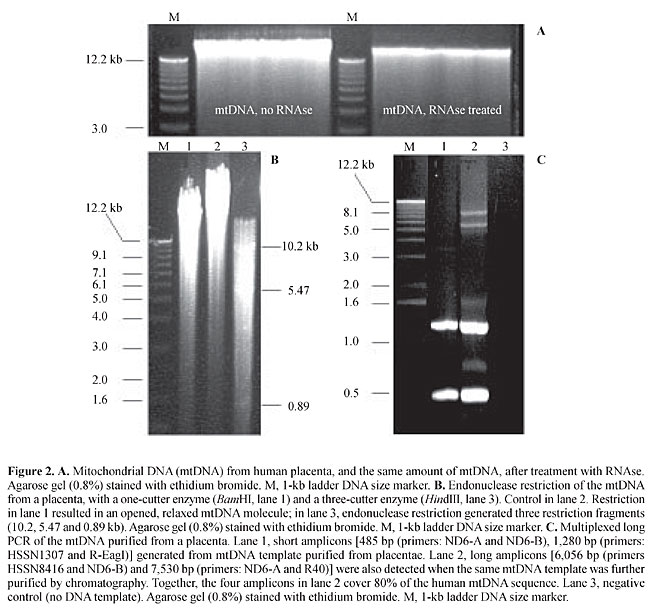
The PCR-amplified, 860-bp fragment was cloned into a pUC18 vector using a Sure CloneÒ Ligation Kit (Pharmacia-Biotech, Uppsala, Sweden) and purified from bacteria using the WizardÒ Miniprep Purification System (Promega, Madison,WI, USA). Sequencing was performed in an ABI PrismÒ377 automatic sequencer (Applied Biosystems). Plasmids containing the 860-bp fragment were digested with restriction endonucleases EcoRI and HindIII; the band corresponding to the released 860-pb fragment, as visualized in an agarose gel, was cut and purified using the Gene Clean Kit (Bio 101 Inc., La Jolla, CA, USA), according to the manufacturer instructions. The purified 860-bp fragment was labeled with [a-32P]-dCTP using the Random Primers DNA Labeling System (Invitrogen, San Diego, CA, USA), according to the manufacturer’s instructions. Hybridization After pre-hybridization of the nylon membrane in hybridization solution [50% phosphate stock solution (0.5 M Na2HPO4, 0.2% (v/v) H3PO4), 7% SDS, 1% BSA and 1 mM EDTA] at 65°C for 1 h, D-probe was added to the nylon membrane. After 12 h hybridization, the membrane was washed twice with buffer 1 (250 ml phosphate stock solution, 1 ml EDTA 0.5 M, pH 8.0, 25 ml SDS 20%, qsp. 500 ml sterile milliQ water) for 30 min at 65°C and twice with buffer 2 (125 ml phosphate stock solution; 1 ml EDTA 0.5 M, pH 8.0; 25 ml SDS 20%; qsp. 500 ml sterile milliQ water), for 30 min. The membrane was treated with 2x SSC and exposed to X-ray film (Kodak, USA) at -80°C for 1 week. RESULTS AND DISCUSSION Average mtDNA yield of the above purification method was 2.5 mg mtDNA/g tissue (without the RNAse treatment step) and 1.5 mg mtDNA/g tissue (with the RNAse treatment step). The quality of the mtDNA preparation was very good, even though some degraded DNA/RNA was seen as a smear in agarose gels (Figure 2); however, this can be viewed as an almost inevitable consequence of the manipulation of a large DNA molecule, which is highly susceptible to shearing forces.
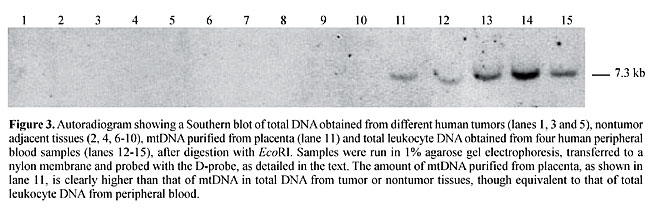
However, the bulk of purified mtDNA migrated as a distinct 16.5-kb band in agarose gels (Figure 2A). There was no apparent sign of contamination with nuclear DNA, since no DNA was seen over the 16.5 kb level. The purified mtDNA yielded fragments of expected sizes after digestion with HindIII (a three-cutter enzyme) and BamHI (a one-cutter enzyme) (Figure 2B); it successfully served as a template in PCRs for amplification of mtDNA sequences (Figure 2C), and it efficiently hybridized to an mtDNA probe (Figure 3). The sizes of the mtDNA fragments generated by both restriction endonucleases and long PCR amplification assays and in the autoradiograms were as would be predicted by the Cambridge mtDNA sequence (1), thus confirming the identity of our purified mtDNA.
In our multiplexed PCRs, shorter amplicons (485-1,280 bp) were efficiently generated from the mtDNA template purified from placenta, with no further purification. However, long amplicons (6,056-7,530 bp) were best detected when the mtDNA template had been further purified by chromatography, under the above multiplex conditions. The amelioration of the competitive inhibition by chromatography indicates the presence of some inhibiting contaminant in the preparation, and imposes a need for further purification of the placental mtDNA, at least for long PCR applications. The purified placental mtDNA hybridized to our mtDNA probe, D-probe, in a much more intense fashion than with total DNA from other tissues, such as solid tumors or their adjacent normal tissues, though as intensely as with total leukocyte DNA from peripheral blood (Figure 3). This behavior is consistent with the view that the placenta purification method provides amounts of mtDNA at least comparable to the amount of mtDNA present in total DNA from peripheral leukocytes, with the advantage that more placenta tissue can be obtained for mtDNA purification than other solid tissue or even blood, at lower cost and raising minimal or no ethical issues. MtDNA yields of the present method are 10-fold higher than those of previously reported ones for mtDNA purification from freshly obtained human cells and tissues such as platelets and placentae, which typically range from 0.05-0.1 mg/g tissue (King and Attardi, 1988; Ausenda and Chomyn, 1996). The almost two-fold difference in yield between nontreated and RNAse-treated mtDNA reflects the transcriptionally active state of the mtDNA molecule. In human cells, transcriptionally active mtDNA can be found complexed with two large RNA strands, each comprising a full-length copy of the mtDNA (Alberts et al., 1994). RNAse digestion of the mtDNA obtained from placentae is thus mandatory if a highly purified mtDNA preparation is needed. A further chromatographic purification step has proven useful for the preparation of the mtDNA to serve as a template in long PCR. ACKNOWLEDGMENTS Research supported by FAPESP (grant No. 97/06229-5) and Genon Ltd. REFERENCES Alberts, B. (ed.), Bray, D., Lewis, J., Raff, M., Roberts, K. and Watson, J.D. (1994). Molecular Biology of the Cell. 3rd edn. Garland Publishing, New York, NY, USA, pp. 393. Anderson, S., Bankier, A.T., Barrell, B.G., de Bruijn, M.H., Coulson, A.R., Drouin, J., Eperon, I.C., Nierlich, D.P., Roe, B.A., Sanger, F., Schreier, P.H., Smith, A.J., Staden, R. and Young, I.G. (1981). Sequence and organization of the human mitochondrial genome. Nature 290: 457-465. Ausenda, C. and Chomyn, A. (1996). Purification of mitochondrial DNA from human cell cultures and placenta. Methods Enzymol. 264: 122-128. Bigger, B., Tolmachov, O., Collombet, J.-M. and Coutelle, C. (2000). Introduction of chloramphenicol resistance into the modified mouse mitochondrial genome: Cloning of unstable sequences by passage through yeast. Anal. Biochem. 277: 236-242. Chomyn, A. (1996). Platelet-mediated transformation of human mitochondrial DNA-less cells. Methods Enzymol. 264: 334-339. Dani, S.U., Dani, M.A.C., Simpson, A.J.G., Pittella, J.E.H., Zullo, S. and Merril, C.R. (1998). Incidência da mutação-deleção mtDNA4977 e quantidade de placas neuríticas e emaranhados neurofibrilares no córtex entorrinal humano. Rev. Psiquiatr. Clin. 25: 118-125. Drouin, J. (1980). Cloning of human mitochondrial DNA in Escherichia coli. J. Mol. Biol. 140: 15-34. Gasnier, F., Rousson, R., Lermé, F., Vaganay, E., Louisot, P. and Gateau-Roesch, O. (1993). Use of percoll gradients for isolation of human placenta mitochondria suitable for investigating outer membrane proteins. Anal. Biochem. 212: 173-178. Hare, J.F., Ching, E. and Attardi, G. (1980). Isolation, subunit composition, and site of synthesis of human cytochrome c oxidase. Biochemistry 19: 2023-2030. King, M.P. and Attardi, G. (1988). Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell 52: 811-819. Reynier, P. and Malthiery, Y. (1995). Accumulation of deletion in mtDNA during tissue aging: Analysis by long PCR. Biochem. Biophys. Res. Commun. 217: 59-67. Zullo, S.J. (2002). In situ PCR of the common human mitochondrial DNA deletion: Is it related to apoptosis? In: Mitochondrial DNA: Methods and Protocols (Copeland, W.C., ed.). Humana Press, Inc., Clifton, NJ, USA. |
|